REVISIÓN |
Medicamenta non mella: los efectos secundarios de los fármacos
María del Carmen Avendaño López
Académica de Número de la Real Academia Nacional de Farmacia.
Recibido el 19 de octubre de 2011.
e-mail: edicion@ranf.com
RESUMEN
En esta revisión se comentan ejemplos representativos de fármacos “sucios”, poniendo de relieve las ventajas e inconvenientes de los efectos secundarios. La biología de sistemas computacional se está utilizando para revelar los mecanismos responsables de estos efectos secundarios y predecir nuevas indicaciones terapéuticas para los viejos fármacos. |
Palabras clave: Efectos secundarios o adversos; Nuevas indicaciones terapéuticas; Biología de sistemas computacional.
ABSTRACT
Medicamenta non mella: the secondary effects of drugs
Representative examples of “dirty” drugs, emphasizing the advantages and drawbacks of their secondary effects, are reviewed. Computational systems biology is being now applied to discover the underlying mechanisms of secondary effects and predict new therapeutic indications for old drugs. |
Keywords: Adverse or secondary effects; New therapeutic indications; Computational systems biology.
1. introducción
Acaban de publicarse los resultados de un estudio a largo plazo, financiado por el National Cancer Institute de los Estados Unidos e iniciado a mediados de los años 70, para determinar la magnitud y el alcance de los efectos adversos provocados por el primer estrógeno sintético: el dietilestilbestrol (DES) (1). Esta noticia nos lleva a reflexionar una vez más sobre los efectos secundarios de los medicamentos, como nos recuerda el aforismo de Plinio el Viejo adoptado como lema por la Real Academia Nacional de Farmacia: Medicamenta non mella.
El dietilestilbestrol se sintetizó por primera vez en 1938 en el grupo que dirigía Sir Robert Robinson, galardonado con el premio Nobel de Química en 1947 (2), y se administró entre los años 40 y 70 a mujeres en gestación basándose en la falsa creencia de que podía evitar riesgos en el embarazo. Millones de mujeres, denominadas “DES daughters”, se expusieron a este compuesto en el útero de sus madres antes de que su uso declinara en los años 50, cuando varios estudios evidenciaran su ineficacia, y de su retirada en 1971 al descubrirse la aparición precoz de adenocarcinomas de vagina y cervix en estas mujeres (3). Estudios posteriores corroboraron la inducción de otras malformaciones vaginales así como problemas en el embarazo (4) sugiriéndose varios mecanismos por los que se producen estos efectos adversos (5). En el mencionado estudio a largo plazo se han puesto de manifiesto efectos adversos como infertilidad, abortos espontáneos, partos a pretérmino, embarazos ectópicos, preeclampsia, menopausia precoz, y mayor riesgo de neoplasia intraepitelial y cáncer de mama, lo que evidencia el alto el riesgo vital que supuso para la descendencia femenina.
A pesar de su triste historia, el dietilestilbestrol ha sido muy útil. Baste recordar que la manipulación de su estructura dio lugar al fármaco antiestrogénico tamoxifeno, utilizado desde finales de los años 70 en la prevención y el tratamiento del cáncer de mama, y a otros fármacos trifeniletilénicos como toremifeno moduladores selectivos de receptores estrogénicos (SERMs). La asociación de la toxicidad a largo plazo que pueden desarrollar estos fármacos con la presencia en su estructura del doble enlace etilénico dio origen a fármacos más seguros para la prevención del cáncer de mama y el tratamiento de la osteoporosis como raloxifeno (Figura 1) (6).

Figura 1.- Dietilestilbestrol y fármacos derivados por manipulación estructural.
2. Los efectos secundarios o adversos
Los fármacos se clasifican, más que por su estructura química, por su acción farmacológica, aunque la mayor parte de ellos producen varias acciones. Cada efecto es consecuencia de un determinado mecanismo de acción que describe la interacción química entre el fármaco y una diana biológica concreta, generalmente una proteína. El efecto principal es el que marca la aplicación terapéutica que se somete a evaluación en los ensayos clínicos, mientras que los efectos secundarios, también denominados efectos adversos, pueden en realidad ser perjudiciales o beneficiosos. El panorama se complica porque el mecanismo de acción de muchos fármacos se desconoce y, además, distintos mecanismos de acción pueden dar lugar a una misma indicación terapéutica. Cada vez es más evidente que existen múltiples vías implicadas en la fisiopatología de las enfermedades y, en consecuencia, está justificado que no se pretendan desarrollar fármacos con acciones únicas puras sino fármacos de acción múltiple. Coloquialmente, los fármacos que pueden interaccionar con varias dianas biológicas diferentes se denominan fármacos “sucios” (dirty drugs). Durante mucho tiempo la industria farmacéutica trató de evitarlos desarrollando fármacos más selectivos a fin de reducir el riesgo de encontrar acciones adversas que, por ser poco frecuentes o de lento desarrollo, se observan muchas veces tras su autorización. Sin embargo, actualmente se admite que, para tener un fármaco eficiente, éste debe interaccionar con múltiples dianas y que muchos fármacos “sucios”, además de utilizarse en terapéutica, pueden ampliar sus indicaciones, en función de sus efectos secundarios.
3. El hallazgo de nuevas indicaciones terapÉuticas basado en la observación de efectos secundarios
Desde el comienzo de la farmacología es frecuente el hallazgo de nuevos usos para los fármacos, a través de la observación y el estudio de los efectos secundarios que producen antes y, fundamentalmente, después de su comercialización. La observación en las sulfamidas antibacterianas de efectos diuréticos e hipoglucemiantes, producidos por inhibición de la enzima anhidrasa carbónica o por bloqueo de los canales de K+ regulados por ATP, dio lugar a diuréticos como hidroclorotiazida, acetazolamida y metazolamida, así como a antidiabéticos orales como glibenclamida y glipizida. La aspirina es otro fármaco clásico para el que se que han encontrado varias indicaciones y hasta es posible que se desarrolle como coadyuvante en el tratamiento de ciertos tipos de cáncer (7). Además, la observación de efectos secundarios inesperados en los inicios de la investigación clínica de un fármaco puede conducir a que éste se desarrolle para una aplicación terapéutica distinta a aquélla para la que fue diseñado. Así, el raloxifeno fracasó como anticonceptivo oral pero se utiliza en el tratamiento de la osteoporosis debido al aumento de densidad ósea que produce; el sildenafilo (Viagra®), utilizado en el tratamiento de la disfunción eréctil, se diseñó en un principio para el tratamiento de la hipertensión arterial y de la angina de pecho, pero en los ensayos clínicos de fase I se comprobó que, aunque tenía un ligero efecto antianginoso, producía una erección prolongada del pene.
Dadas las dificultades encontradas en los últimos años para introducir fármacos innovadores, el descubrimiento de nuevos usos para viejos fármacos se ha convertido en una gran promesa de nuevas opciones terapéuticas. Curiosamente, veremos que estos descubrimientos no son fruto de la casualidad ni del trabajo de los farmacólogos, sino de los informáticos (8).
4. Problemas y ventajas de los fÁrmacos “sucios”
Los antipsicóticos son representativos de la problemática de los fármacos “sucios”. Su historia comienza con la clorpromazina (Figura 2); introducida en la clínica en los años 50 fue el primer fármaco con acción antipsicótica y neuroléptica, suprimió la terapia electroconvulsiva y las técnicas quirúrgicas como la lobotomía, y eliminó prácticamente el internamiento de estos enfermos en hospitales psiquiátricos (9). Su efecto principal se produce al antagonizar receptores de serotonina por lo que puede usarse en el tratamiento del “síndrome serotoninérgico” caracterizado por un exceso de actividad serotoninérgica, consecuencia de sobredosis de determinados fármacos o de interacciones medicamentosas (10). Sin embargo, al ser un fármaco “sucio”, actúa también sobre otros receptores del sistema nervioso central produciendo efectos anticolinérgicos (antiemético, reductor de la ansiedad, hipotensor y sedante), antidopaminérgicos (por los que puede causar síntomas extrapiramidales como acatasia), antihistamínicos y antiadrenérgicos. Un análogo de clorpromazina, de características parecidas, pero bastante más potente, es perfenazina, utilizada en clínica desde los años 50 (11). Los efectos indeseables de ambos fármacos promovieron la aparición en el mercado de los antipsicóticos de segunda generación (atípicos), un grupo heterogéneo de compuestos no relacionados entre sí que poseían un mecanismo de acción diferente al de los antipsicóticos típicos y parecían ser mejor tolerados. Muchos de ellos actúan sobre receptores de serotonina y de dopamina (12).
La clozapina se lanzó a finales de los años 50 y se considera el prototipo de antipsicótico atípico. Posee mayor eficacia y menores efectos extrapiramidales (temblores y parkinsonismo) que los antipsicóticos típicos; sin embargo es un fármaco que posee muchos efectos adversos como fuerte sedación, aumento de peso, hipertrigliceridemia, etc. A principios de los años 70 se asoció a una serie de casos de agranulocitosis con resultado de muerte, por lo que se retiró del mercado en muchos países. Sin embargo, debido a su eficacia, se volvió a autorizar a finales de los 80 como fármaco de reserva en la esquizofrenia y otros síntomas psicóticos instaurando un protocolo de control hematológico. En 1993 la FDA aprobó la risperidona, en 1996 la olanzapina, y en 1997 la quetiapina. La risperidona tiene pocos efectos extrapiramidales e interacciona con receptores D2 y 5-HT2A, junto con otros receptores serotoninérgicos. La olanzapina es un análogo de clozapina que posee una mayor afinidad por el receptor de serotonina 5-HT2 que por el de dopamina D2. La actividad antagonista que ejerce sobre este último se asocia a sus efectos extrapiramidales y, además, puede producir sedación por ser antagonista de los receptores de histamina H1. La quetiapina actúa como antagonista de receptores 5-HT2A y de receptores dopaminérgicos postsinápticos. Además de mostrar varios de los efectos secundarios de los antipsicóticos, incrementa la prolactina por encima de las tasas normales, lo que podría provocar la aparición de tumores no cancerígenos en la glándula pituitaria (13). Otro antipsicótico atípico que consiguió ser aprobado por la FDA en 2002 fue el aripiprazol que, aunque como otros antipsicóticos atípicos muestra un perfil antagonista del receptor 5-HT2A, difiere en que sus efectos parecen ser la consecuencia de su actividad agonista parcial de receptores D2 y del receptor 5-HT1A.
En el año 2005 se publicó que la quetiapina y otros antipsicóticos atípicos no eran más eficaces en el tratamiento de este síndrome que el antipsicótico de primera generación perfenazina (14), lo que fue refutado en parte a través de ensayos auspiciados por las compañías farmacéuticas que los fabricaban. En resumen, estamos ante un grupo de fármacos cuya relación eficacia/toxicidad, debido a sus múltiples mecanismos de acción, es muy variable.
Ya se ha comentado que la tendencia a seleccionar para su desarrollo fármacos “limpios”, quizás un objetivo imposible, ha ido desdibujándose de tal modo que en los últimos años han adquirido valor los fármacos de acción dual e incluso múltiple. Tomemos como ejemplo los antidepresivos. La comercialización de los que actúan inhibiendo selectivamente la recaptación de serotonina (SSRIs) como la fluoxetina (el famoso Prozac®, Figura 3), fue un gran éxito porque estos fármacos actúan exclusivamente sobre las vías serotoninérgicas, mientras que los antidepresivos comercializados con anterioridad vieron limitada su utilidad clínica por su acción dual ya que en su mayoría actúan sobre las vías serotoninérgicas y noradrenérgicas. Entre ellos se encuentran los antidepresivos tricíclicos (TCAs) como imipramina y amitriptilina y los inhibidores de monoaminooxidasa (MAOIs) como tranilcipromina.

Figura 2.- Algunos ejemplos de fármacos antipsicóticos típicos y atípicos.
Posteriormente, se comprobó que los SSRIs son menos eficaces que los TCAs y los MAOIs, y se despertó el interés por el descubrimiento y desarrollo de nuevos antidepresivos de acción dual que fuesen mejor tolerados que los ya existentes. Así, aunque los antidepresivos los SSRIs de acción única son todavía los más prescritos, han surgido con fuerza antidepresivos de acción dual inhibidores de la recaptación de serotonina y de noradrenalina como venlafaxina, mirtazapina y duloxetina, que poseen menos efectos secundarios indeseables que los antiguos antidepresivos de acción dual TCAs y MAOIs (15). La mayor eficacia de los antidepresivos de acción dual se apoyó en estudios que demostraban la reaparición de los síntomas depresivos si tras el tratamiento de fármacos serotoninérgicos se daba una dieta que disminuyera los niveles de serotonina, ocurriendo lo mismo en pacientes tratados con fármacos adrenérgicos cuando se rebajaban los niveles de noradrenalina. Por el contrario, si en los primeros se disminuían los niveles de noradrenalina o en los segundos los de serotonina, los síntomas no reaparecían de forma significativa. Por tanto, la actuación conjunta sobre los sistemas de serotonina y noradrenalina puede conducir a una mejor actividad antidepresiva.

Figura 3.- Ejemplos de fármacos antidepresivos.
En el campo de los antitumorales mencionaremos a imatinib, más conocido por su nombre comercial Glivec® (Figura 4). Fue en su momento un fármaco muy relevante por ser el primer antitumoral diseñado para inhibir la actividad de la diana causante de la leucemia mieloide crónica (16), la tirosina cinasa de Abelson (ABL), que se encuentra desregulada y formando un híbrido oncogénico con la proteína de resistencia de cáncer de mama BCR (por ello se denomina cinasa BCR-ABL) (17). Recordemos que las cinasas fosforilan grupos hidroxilo de los aminoácidos tirosina, serina o treonina de determinadas proteínas por transferencia de un grupo fosfato desde el trifosfato de adenosina (ATP) produciendo la activación de aquellas.
El mismo mes de ser aprobado por la agencia norteamericana FDA (mayo de 2001), la revista TIME lo definió como “la bala mágica para curar el cáncer”, siendo el paradigma del “diseño racional de fármacos” (18). Imatinib se une a una conformación inactiva de la cinasa ABL mimetizando al ATP, pero en su diseño se partió de compuestos que habían demostrado ser inhibidores de otra cinasa de serina-treonina: la proteína cinasa C (PKC). La estructura de estos prototipos se optimizó hasta encontrar inhibidores duales de las cinasas PKC y ABL y, tras solventar algunos problemas relacionados con un metabolismo inadecuado, se sintetizó un buen número de análogos hasta encontrar un inhibidor selectivo ABL que finalmente se modificó para resolver problemas de farmacocinética. Más tarde se comprobó que imatinib no es la “bala mágica” que interacciona exclusivamente con la cinasa BCR-ABL sino que inhibe además otras cinasas. Frente a lo que pudiera pensarse, esta característica no le ha quitado valor, sino que ha aumentado sus indicaciones terapéuticas. Entre las cinasas inhibidas por imatinib se encuentran la receptora del factor de crecimiento derivado de plaquetas (PDGF), que tiene un papel importante en el desarrollo de glioma, cáncer de próstata, y cáncer de células pequeñas de pulmón, por lo que se investiga su uso en esas patologías. También inhibe la cinasa c-KIT (cell tyrosin kinase) (19) que se encuentra mutada en el estroma gastrointestinal (20) inhibiendo la fosforilación del factor de crecimiento hematopoyético. Por ello fue aprobado por la FDA en el año 2002 para el tratamiento de este tipo de tumores.

Figura 4.- Imatinib.
Comentario aparte merecen las estatinas (Figura 5) que, a pesar de su corta historia, se utilizan por cientos de miles de pacientes como hipocolesterolémicos a fin de prevenir la ateroesclerosis y las patologías cardiovasculares. Su descubrimiento también fue relevante porque primeramente se identificó la diana biológica que debía inhibirse: la hidroximetil glutaril coenzima A (HMG-CoA) reductasa, una enzima que cataliza uno de los pasos de la biosíntesis del colesterol. Posteriormente, a través del cribado masivo de muchos fármacos y extractos naturales, se encontró una molécula activa aislada del hongo Penicillium citrinum a la que se denominó mevastatina, pero su desarrollo tuvo que interrumpirse debido a su elevada toxicidad. En 1979 se aisló de Aspergillus terreus, lovastatina y, posteriormente, de Nocardia autotrophica, pravastatina. La primera estatina sintética fue fluvastatina comercializada en 1994, a la que siguió simvastatina, obtenida a partir de un precursor producido también por Aspergillus terreus; atorvastatina, también totalmente sintética (21), es la más prescrita. Aunque estudios de fase IV han motivado la retirada de algún miembro de esta familia que poseía graves efectos secundarios, el desarrollo de nuevas estatinas ha continuado hasta la actualidad.
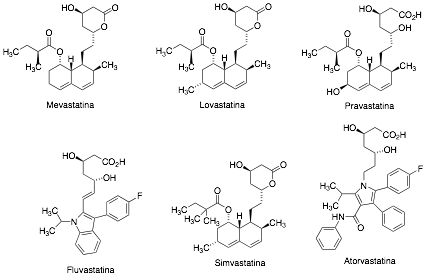
Figura 5.- Ejemplos de estatinas.
Gran parte de su éxito se debe a que estos fármacos no se limitan a inhibir la enzima HMG-CoA reductasa sino que poseen otras muchas acciones (22) a través de mecanismos independientes de su mecanismo de acción principal (23). Producen una disminución de las moléculas de adhesión, disminuyen la oxidación del LDL en el espacio subendotelial (24), la actividad metabólica de los macrófagos en las placas de ateroma (25), la proliferación celular en el músculo liso vascular, la síntesis de endotelina-1 (26), la agregación plaquetaria y la actividad del inhibidor del activador de plasminógeno-1 (PAI-1) previniendo la formación de trombos (27). Disminuyen también los marcadores sistémicos de inflamación (28) y la respuesta inmunológica (29) y estimulan la sintetasa de óxido nítrico endotelial (30). Uno de sus efectos más importantes es la reducción de la proliferación y viabilidad de células tumorales y el control de la metástasis (31-33). Además, se presume que las estatinas tienen un efecto más amplio debido a que pueden inducir una disminución del crecimiento tumoral por inhibir la activación de las proteínas Ras, Rho, p21 y p27, por activar caspasas, disminuir las proteínas bcl-2, e inhibir la selectina E y la metaloproteinasa 9.
Resulta llamativo que algunos fármacos considerados “malditos” por sus efectos secundarios se sigan utilizando en terapéutica para diversos propósitos. El ejemplo más conocido es la talidomida (Figura 6) comercializada entre los años 1958 y 1963 como sedante y calmante de las náuseas en la hiperémesis gravídica durante los tres primeros meses de embarazo. Tras obtener un éxito clamoroso produjo una alarma mundial cuando se supo que tenía efectos teratogénicos que habían provocado miles de nacimientos de bebés afectados de focomelia, una anomalía congénita caracterizada por la carencia o acortamiento de las extremidades. Como consecuencia, muchos países establecieran paulatinamente un control estricto de los medicamentos que pretenden comercializarse para garantizar su eficacia y su baja toxicidad (34). Estudios posteriores a su retirada indicaron que el enantiómero R producía el efecto sedante y carecía de teratogenicidad, siendo el S el responsable de este efecto (35) y a partir de ese momento se prestó atención a la evaluación de ambos enatiómeros en el desarrollo de fármacos quirales. Podría pensarse que el enantiómero R puro de la talidomida podría utilizarse sin problemas, pero lo cierto es que, dada la acidez del átomo de hidrogeno del centro estereogénico en posición a a un grupo carbonilo, ambos enantiómeros se interconvierten tanto si la talidomida se administra por vía oral como intravenosa (36). Tras el descubrimiento casual de sus beneficios en los pacientes afectados de lepra este fármaco siguió comercializándose para esta indicación, pero su verdadera rehabilitación comenzó a finales de los años 90 cuando se observó que poseía propiedades inmunomoduladoras, antiinflamatorias, y antiangiogénicas a través de varios mecanismos como la modulación de la síntesis de citocinas (especialmente del factor de necrosis tumoral alfa, TNFa) (37) y la inhibición de la fagocitosis realizada por los linfocitos polimorfonucleares en las células T, debido a la mayor producción de interleucina 2, IL-2 (38). De hecho, la talidomida se vuelve a utilizar como terapia alternativa o de segunda elección bajo un estricto control médico en algunas afecciones dermatológicas como el prúrigo nodular y actínico, el lupus eritematoso, las úlceras aftosas asociadas a la infección del virus VIH, y la estomatitis aftosa recurrente, así como en algunos tipos de cáncer. También se obtienen buenos resultados en enfermedades inflamatorias intestinales como la enfermedad de Crohn, la artritis reumatoide e incluso en la insuficiencia cardiaca avanzada. En resumen, la talidomida promete ser un fármaco con múltiples aplicaciones siempre y cuando se siga una adecuada vigilancia para la identificación temprana de sus efectos colaterales (39).
Además, un análogo estructural de talidomida: lenalidomida (EM 12 ó CC-4047) se aprobó en el año 2008 por la Agencia Europea del Medicamento (EMA) para el tratamiento del mieloma múltiple, recomendándose su utilización en pacientes refractarios a otros tratamientos (40). Este nuevo fármaco posee propiedades antiangiogénicas a través del bloqueo de la migración y adhesión de células endoteliales y de la formación de microvasos, propiedades proeritropoyéticas, e inmunomoduladoras (al potenciar la inmunidad celular mediada por linfocitos T e inhibir la producción de citocinas proinflamatorias como TNF-α e IL-6). Al igual que la talidomida, los dos enantiómeros de lenalidomida se intercambian muy fácilmente entre sí (41, 42), aunque la configuración de algunos análogos de lenalidomida en los que se ha introducido un grupo alquilo o arilo en la posición 4, es más estable (43). Debido a que podría provocar malformaciones fetales por su similitud con la talidomida, no debe administrarse a mujeres embarazadas, recomendándose evitar el embarazo desde el mes previo al inicio del tratamiento hasta 4 semanas después de su finalización.
 .
.
Figura 6.- Talidomida y análogos.
5. Cómo empiezan a descubrirse los secretos que esconden los fÁrmacos “sucios”
Los efectos secundarios son respuestas fenotípicas del organismo cuando éste se trata con un determinado fármaco. Aunque la industria farmacéutica está muy interesada en predecirlos (44, 45), los estudios preclínicos que comúnmente tratan de evaluar los efectos atribuidos a un mecanismo de acción aceptado o supuesto no son suficientemente predictivos en este sentido, lo que explica el gran número fracasos que se obtienen en los ensayos clínicos. Por ello, se están desarrollando nuevos métodos computerizados (46) para descubrir todas las dianas biológicas con que interaccionan los fármacos y sus correspondientes vías de señalización utilizando células u organismos vivos como modelos humanos (47, 48). Los ensayos robotizados “protein fragment complementation assays” (PCAs) permiten visualizar un amplio espectro de estas vías mediante el análisis de las interacciones proteína-proteína; la actividad de una proteína se determina fusionándola con un fragmento de otra que puede visualizarse por fluorescencia (“reporter protein”) de tal forma que, si dos proteínas se expresan en una célula viva e interaccionan para formar un complejo, se puede obtener una señal detectable. Si estos ensayos se combinan con el cribado de fármacos de alto rendimiento, es posible determinar la dinámica de los complejos que resultan de las respuestas celulares a los fármacos que activan o inhiben una vía determinada. De esta forma, se pueden identificar cuáles son los fenotipos que esconden los fármacos y cuáles son los mecanismos bioquímicos responsables de sus efectos terapéuticos y de su toxicidad.
En el año 2006 se publicó un interesante trabajo en el que se estudiaron 107 fármacos pertenecientes a 6 indicaciones terapéuticas distintas frente a 49 “PCA reporters” en 10 procesos celulares diferentes (49) y aquellos se agruparon según su estructura y sus dianas biológicas. Además de reproducirse las relaciones estructura-actividad ya conocidas, se vio que el anticolesterolémico fenofibrato, el antihistamínicao cinarizina, el antihelmíntico niclosamida, y el antidepresivo ISRS sertralina poseían una actividad antiproliferativa en una línea celular de carcinoma de próstata y en otros cuatro tipos de cáncer (Figura 7). También se pudieron identificar en este estudio 25 PCAs con capacidad para predecir dicha actividad y aunque en la proliferación celular están implicadas numerosas vías, dado que estos ensayos están diseñados para analizar los efectos en vías concretas y los compuestos que afectan a las mismas vías de señalización producen fenotipos similares, se pudo también proponer el fenotipo antiproliferativo.

Figura 7.- Fármacos en los que se descubrió una actividad antiproliferativa.
Obviamente, la predicción de interacciones desconocidas entre un fármaco ya comercializado y una diana biológica puede utilizarse para proponer una nueva indicación terapéutica, con la ventaja de que ya se ha comprobado su margen de seguridad por lo que si se descubre en ellos una nueva aplicación clínica ésta puede aprobarse más rápidamente que si se trata de un nuevo fármaco que normalmente requiere más de 10 años. Peer Bork, coordinador de un equipo de investigación de la Unidad de Biología Estructural y Computacional del Laboratorio Europeo de Biología Molecular (EMBL) que investiga en esta dirección, afirma que la similitud de los efectos secundarios que producen diferentes fármacos indica que estos efectos se originan como consecuencia de las interacciones con una misma diana molecular, revelando la base molecular de aquellos, lo que puede dar lugar a la utilización de fármacos comercializados en el tratamiento de enfermedades para los que no se habían desarrollado. Su equipo desarrolló un método informático para un estudio con 746 fármacos descubriéndose que 261, además de su conocida actividad farmacológica, poseían efectos secundarios similares por lo que podrían interaccionar con dianas moleculares inesperadas. Se seleccionaron 20 de estos 261 fármacos para estudiarlos experimentalmente, y se encontró que 13 se unían a las dianas que se habían predicho. De estos 13 fármacos se seleccionaron 9 para estudiar experimentalmente sus efectos a nivel celular, y todos ellos presentaron la actividad esperada (50). Este grupo también ha utilizado los efectos secundarios como una importante fuente de información de los fenotipos que producen los fármacos, esencial para determinar los mecanismos de acción y para desarrollar fármacos personalizados. Para ello, ha desarrollado la herramienta informática SIDER (“side effect resource”), disponible para la investigación académica (51), que conecta 888 fármacos con 1450 efectos secundarios (52).
Confiamos haber puesto de manifiesto el gran potencial que posee la predicción de los posibles beneficios de los efectos secundarios de los fármacos para abrir la puerta a su utilización en indicaciones terapéuticas diferentes a las ya aprobadas (53).
6. REFERENCIAS
1. Hoover, R. N., Hyer, M., Pfeiffer, R. M. et al. (2011). Adverse Health Outcomes in Women Exposed In Utero to Diethylstilbestrol. N. Engl. J. Med., 365, 1304-1314.
2. Dodds, E. C., Goldberg, L., Lawson, W., & Robinson, R. (1938). Estrogenic activity of certain synthetic compounds. Nature, 141, 247-248.
3. a).Herbst, A. L., Ulfelder, H., & Poskanzer, D. C. (1971). Adenocarcinoma of the vagina: association of maternal stilbestrol therapy with tumor appearance in young women. N. Engl. J. Med., 284, 878-881. b).Ulfelder, H. (1980). The stilbestrol disorders in historical perspectiva, Cancer. 45, 3008-3011.
4. a).Herbst, A. L., Kurgan, R. J., & Scully, R. E. (1972). Vaginal and cervical abnormalities after exposure to stilbestrol in utero. Obstet. Gynecol., 40, 287-298. b).Barnes, A. B., Colton, T., Gundersen, J. et al. (1980). Fertility and outcome of pregnancy in women exposed in utero to diethylstilbestrol, N. Engl. J. Med. 302, 609-613.
5. Diamanti-Kandarakis, E., Bourguigon, J. P., Giudice, L. C. et al. (2009). Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr. Rev., 30, 293-342.
6. Avendaño, C., & Menéndez, J. C. (2008). Medicinal Chemistry of Anticancer Drugs, ed. Elsevier, pág. 58-62.
7. Rothwell, P. M., Fowkes, G. R., Belch, J. F., Ogawa, H., Warlow, Ch. P., & Meade, T. W. (2010). Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. The Lancet, 377, 31-41.
8. Shaughnessy, A. F. (2011). Old drugs, new tricks. British Medical Journal, 9, 342, d741.
9. Healy, D. (2004). The creation of Psychopharmacology. Harvard University Press, pág. 37-73.
10. Boyer, E. W., & Shannon, M. (2005). The serotonin syndrome. New England Journal of Medicine, 352 , 1112–20.
11. Rees, L. (1960). Chlorpromazine and allied phenothiazine derivatives. British Medical Journal, 2, 522–225.
12. Leucht, S., Corves, C., Arbter, D., Engel, R. R., Li, Ch., & Davis, J. M. (2009). Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. The Lancet, 373, 31-41.
13. Szarfman, A., Tonning, J., Levine, J., & Doraiswamy, P. (2006). Atypical antipsychotics and pituitary tumors: a pharmacovigilance study. Pharmacotherapy, 26, 748-758.
14. Lieberman, J. A., Stroup, T. S., McEvoy, J. P., Swartz, M. S., Rosenheck, R. A., Perkins, D. O., Keefe, R. S. E., Davis, S. M., Davis, C. E., Lebowitz, B. D., Severe, J., & Hsiao, J. K. (2005). Effectiveness of Antipsychotic Drugs in Patients with Chronic Schizophrenia. New England Journal of Medicine, 353, 1209 -1223.
15. Rakesh, J. (2004). Single-Action Versus Dual-Action Antidepressants, Journal of Clinical Psichiatry, 6, 7-11.
16. Deininger, M. W., & Druker, B. J. (2003). Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacological Reviews, 55, 401–23.
17. Manley, P. W., Cowan-Jacob, S. W., & Mestan, J. (2005). Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochimica et Biophysica Acta, 1754, 3-13.
18. Druker, B. J., & Lydon, N. B. (2000). Lessons learned from the development of an Abl tyrosine kinase inhibitor for chronic myelogenous leucemia, Journal of Clinical Investigation, 105, 3-7.
19. Guilhot, F. (2004). Indications for Imatinib Mesylate Therapy and Clinical Management. The Oncologist, 9, 271–281.
20. Hirota, S., Isozaki, K., Moriyama, Y., Hashomoto, K., Nishida, T., Ishoguro, S., Kawano, K., Hanada, M., Kurata, A., Takeda, M., Muhammad Tunio, G., Matsuzawa, Y., Kanakura, Y., Shinomura, Y., & Kitamura, Y. (1998). Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science, 279: 577-80.
21. Jonson, D. S., & Li, J. J. (2007). The art of Drug Synthesis, Wiley-Interscience.
22. Davignon, J., & Laaksonen, R. (1999). Low-density lypoprotein independent effects of statins. Current Opinion in Lipidology, 10, 543-559.
23. Singh, V., & Deedwania, P. (2008). Expanding roles for atorvastatin. Drugs of Today, 44, 455-471.
24. Aviram, M., Dankner, G., Cogan, U., Hochgraf, E., & Brook, J. G. (1992). Lovastatin inhibits LDL oxidation and alters its fluidity and uptake by macrophages: in vitro and in vivo studies. Metabolism, 41, 229-235.
25. Toschi, V., Gallo, R., Lettino, M., Fallon, J. T., Gertz, S., Fernández-Ortiz, A., Chesebro, J. H., Badimon, L., Nemerson, Y., Fuster, V., & Badimon, J. J. (1997). Tissue factor modulates the thrombogenicity of human atherosclerotic plaque. Circulation, 95, 594-599.
26. Hernández-Perera, O., Pérez-Sala, D., Navarro-Antolín, J., Sánchez-Pascuala, R., Hernández, G., Díaz, C., & Lamas, S. (1998). Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endotelial nitric oxide synthase in vascular endotelial cells. Journal of Clinical Investigation, 101, 2711-2719.
27. Fenton, J. W., & Shen, G. X. (1999). Statins as cellular antithrombotics. Haemostasis, 29, 166-169.
28. Jialal, I., Stein, D., Balis, D., Adams-Huet, B., & Devaraj, S. (2001). Effect of hydroxymethyl glutaryl coenzyme A reductase inhibitor therapy on high sensitive C-Reactive protein levels. Circulation, 103, 1933-1935.
29. Kwak, B., Mulhaupt, F., & Mach, F. (2000). Statins as a newly recognized type of immunomodulator. Natural Medicine, 6, 1399-1402.
30. Laufs, U., La Fata, V., Plutzky, J., & Liao, J. K. (1998). Upregulation of endotelial nitric oxide synthase by HMG-CoA reductase inhibitors. Circulation, 97,1129-1135.
31. Hindler, K., Cleeland , C. S., Rivera, E., & Collard, C. D. (2006). The role of statins in cancer therapy. The Oncologist, 11, 306-315.
32. Sassano, A., & Platanias, L. C. (2008). Statins in tumor suppression. Cancer Letters, 260, 11-19.
33. Fritz, G. (2009). Targeting the mevalonate pathway for improved anticancer therapy. Current Cancer Drug Targets, 9, 626-638.
34. Rouhi, M. (2005). Top Pharmaceuticals: Thalidomide, Chemical & Engineering News, 83,122-123.
35. Blaschke, G., Kraft, H. P., Fickentscher, K., & Kohler, F. (1979). Chromatographic separation of racemic thalidomide and teratogenic activity of its enantiomers (traducción). Arzneimittelforschung, 29, 1640–1642.
36. Eriksson, T., Bjorkman, S., & Hoglund, P. (2001). Clinical pharmacology of thalidomide. Eur. J. Clin. Pharmacol. 57, 365–376.
37. Sampaio. E. P., Sarno, E. N., Galilly, R., Cohn, Z. A., & Kaplan, G. (1991). Thalidomide selectively inhibits tumor necrosis factor (α) production by stimulated human monocytes. J. Exp. Med., 173, 699-703.
38. Corral, L. G., & Kaplan, G. (1999). Immunomodulation by thalidomide and thali- domide analogues. Annals of the Rheumatic Diseases, 58, 1107-1113.
39. González Infante, E. (2001). Desarrollo químico y galénico de la talidomida como medicamento huérfano, Tesis Doctoral UCM, ISBN: 84-669-2012-9.
40. Dimopoulus, M., Spencer, A., Attal, M., Prince, H. M., Harousseau, J. L., Olesnyckyj, M., Yu, Z-, Patin, J., Zeldis, J. B., & Knight, R. D. (2007). Lenalidomide plus dexamethasone for relapsed or refractory múltiple myeloma. N. Engl. J. Med., 357, 2123-32.
41. Schmahl, H. J., Nau, H., & Neubert, D. (1988). The enantiomers of the teratogenic thalidomide analogue EM 12: Chiral inversion and plasma pharmacokinetics in the marmoset monkey. Archives of Toxicology, 62, 200–204.
42. Teo, S. K., Chen, Y., Muller, G. W., Chen, R. S., Thomas, S. D., Stirling, D. I., & Chandula, R. S. (2003). Chiral inversion of the second generation of CC-4047 in human plasma and phosphate-buffered saline. Chirality, 15, 348–351.
43. Yamada, T., Okada, T., Sakaguchi, K., Ohfune, Y., Ueki, H., & Soloshonok, V. A. (2006). Efficient asymmetric syntheses of novel 4-substituted and configurationally stable analogues of thalidomide. Organic Letters, 8, 5625–5628.
44. Krejsa, C. M., Horvath, D., Rogalski, S. L., Penzotti, J. E-, Mao, B., Barbosa, & F., Migeon, J. C. (2003). Predicting ADME properties and side effects: the BioPrint approach, Current Opinion in Drug Discovery, 6, 470–480.
45. Bender, A., Scheiber, J., Glick, M., Davies, J. W., Azzaoui, K., Hamon, J., Urban, L., Whitebread, S., & Jenkins, J. L. (2007). Analysis of pharmacology data and the prediction of adverse drug reactions and off-target effects from chemical structure. Chem. Med. Chem., 6, 861-873.
46. Chua, H. N., & Roth, F. P. (2011). Discovering the Targets of Drugs Via Computacional Systems Biology. J. Biol. Chem., 286, 23653-23658.
47. Fliri, A. F., Loging, W. T., & Volkmann, R. A. (2007). Analysis of system structure-function relationships. Chem. Med. Chem., 12, 1774-1782.
48. Kaletta, T., & Hengartner, M. O. (2006). Nature Rev. Drug Discovery, 5, 387–398.
49. MacDonald, M. L-, Lamerdin, J., Owens, S., Keon, B. H., Bilter, G. K., Shang, Z., Huang, Z., Yu, H., Dias, J., Minami, T., Michnick, S. W., & Westwick, J. K. (2006). Identifying off-target effects and hidden phenotypes of drugs in human cells. Nature Chemical Biology, 2, 329-337.
50. Campillos, M., Kuhn, M., Gavin, A. C., Jensen, L. J., & Bork, P. (2008). Drug target identification using side-effect similarity. Science, 321, 263-266.
51. http://sideeffects.embl.de.
52. Kuhn, M., Campillos, M., Letunic, I., Jensen, I., Jensen, L. J., & Bork, P. (2010). A side effect resource to capture phenotypic effects of drugs. Molecular Systems Biology, 6, 343.
53. Owens, J. (2006). Dirty drugs’ secrets uncovered. Nature Rev. Drug Discovery 5, 542-553.