ARTÍCULO |
Efectos cardioprotectores de reducción de tamaño del infarto de miocardio y prevención del remodelado ventricular post-infarto del agonista del receptor de esfingosina-1-fosfato: estudio experimental en un modelo porcino de isquemia-reperfusión
Carlos García Santos-Gallego*
*e-mail: carlosgsantos@yahoo.es
An. Real Acad. Farm. Vol. 80, Nº 3 (2014), pag. 694-719
Premio “Real Academia Nacional de Farmacia” en el Concurso Científico 2013 de la Real Academia Nacional de Farmacia.
resumen
La isquemia miocárdica induce fenómenos de muerte celular por necrosis y apoptosis. La apoptosis es un proceso que requiere energía (dependiente de oxígeno y ATP), por lo que -aunque los genes proapoptóticos se activen durante la isquemia- el programa apoptótico no se lleva a cabo hasta la reperfusión (cuando el oxígeno está nuevamente disponible para sintetizar ATP). La esfingosina 1-fosfato es un fosfolípido presente en el cuerpo con propiedades antiapoptóticas ya demostradas. Fingolimod es el único agonista sintético del receptor de esfingosina 1-fosfato (S1P-R) que está aprobado para uso en pacientes (en concreto, para esclerosis múltiple). Nuestra hipótesis consiste en que la activación del receptor S1P-R mediante el tratamiento con Fingolimod durante el periodo isquémico del infarto agudo de miocardio disminuye la apoptosis, aumenta el miocardio preservado, reduce el tamaño de infarto y mitiga el remodelado del ventrículo izquierdo en un modelo porcino de isquemia-reperfusión (I-R). Se indujo isquemia-reperfusión en cerdos Yorkshire mediante la oclusión percutánea de la arteria coronaria descendente anterior en su segmento proximal durante 60 minutos. Los animales fueron aleatorizados a recibir Fingolimod o salino (grupo control). Los animales fueron evaluados con resonancia magnética cardiaca (RM), ecocardiografía 3D, técnicas de hemodinámica invasiva, histología y análisis por Western blot. En un estudio inicial, se observó los cerdos tratados con Fingolimod presentaban a las 24 horas post-IAM menos apoptosis en el miocardio isquémico y más activación de proteínas cardioprotectoras y anti-apoptóticas que los cerdos del grupo control. En un estudio a largo plazo, la RM cardiaca una semana post-infarto (IAM) mostró que, a pesar de tener un tamaño similar de área en riesgo en el miocardio en ambos grupos, Fingolimod redujo significativamente el tamaño del infarto y mejoró la función sistólica del ventrículo izquierdo (VI). Estos efectos cardioprotectores seguían asimismo presentes un mes post-IAM, pues los cerdos tratados con Fingolimod presentaban menor tamaño de IAM, mejor función sistólica de VI y menor remodelado cardiaco a nivel anatómico (expresado como menos dilatación de VI, menor masa de VI y menor pérdida de la morfología normal del VI). Adicionalmente, Fingolimod mitigó el desarrollo del remodelado adverso del ventrículo izquierdo a nivel histológico (una disminución de la hipertrofia de los cardiomiocitos y de la fibrosis intersticial) y molecular (menor activación de Akt y de ERK1/2). Finalmente, el grupo Fingolimod presentaba asimismo menor activación neurohormonal (medida como menores niveles de catecolaminas). La activación precoz de S1P-R mediante el tratamiento con Fingolimod previo a la reperfusión provoca efectos cardioprotectores beneficiosos, incrementa el miocardio salvado, reduce el tamaño de infarto, mejora la función sistólica y la mecánica miocárdica del VI y mitiga el remodelado cardiaco adverso post-IAM. |
Palabras clave: Infarto de miocardio; daño por isquemia-reperfusión; remodelado del VI; esfingosina 1-fosfato; miocardiopatía; apoptosis; farmacología.
abstract
Cardioprotective effects of reduction in size of the myocardial infarction and prevention of the ventricular remodeling post-infarction sphingosine‐1‐phosphate receptor agonist: an experimental study in a porcine model of isquemia‐reperfusion
Myocardic ischemia induces cell death due to necrosis or apoptosis. Apoptosis requires energy (obtained from oxygen and ATP). Th Although proapoptotic genes are activated during ischemia the apoptotic programm does not take place until re-perfusion (when oxygen is again available to synthesize ATP). Sphingosine -1-phosphate is a phospholipid with known antiapoptotic properties. Fingolimod is the only synthetic agonist known of spingosine-1-phosphate receptor ( S1P-R) aproved for use in patients ( with multiple sclerosis). Our hypotheis is that activation of S-1-P R with Fingolimod during the period of ischemia of acute myocardic infartion decreases apoptosis, reduce the size and the process of remodeling of left ventricle in a porcine model of ischemia-reperfusion ( I-R). Thus, I-R was induced in Yorkshire pigs by oclusión of coronary ateria in its proximal segment during 60 min. and treated or not with Fingolimod. Animals were evaluated by cardiac magnetic resonance (RM), ecocardiography 3D, hemodynamic tecniques, histology and Western blot analysis. Pigs treated with Fingolimod after 24 hr showed lower apoptosis of the myocardium and activation of antiapototic proteins Tan control group. In a long term study Fingolimod reduced the size of infartion and improved sustolic function of left ventricle. In addition Fingolimod decreased cardiomyocites hypertrophy and activation of molecular parameters as Akt and ERK 1/2. We conclude that early activation of S1P-R by Fingolimod reduces the size of infartion improves systolic function and mitigates adverse cadiac remodelling . |
Keywords: Myocardial infarction; ischemia-reperfusion; Sphingosine -1-phosphate; myocardiopathy; apoptosis; pharmacology.
1. INTRODUCcióN
La reperfusión precoz mediante cateterismo cardiaco/angioplastia coronaria o mediante trombolisis es el método más utilizado actualmente en el tratamiento de pacientes con infarto agudo de miocardio (IAM). La reperfusión temprana reduce con éxito el tamaño de infarto, preserva la función cardiaca y disminuye la morbilidad y la mortalidad (1). Desafortunadamente, la reperfusión no siempre es suficiente para conseguir la recuperación adecuada del miocardio. De hecho, la reperfusión por sí misma puede paradójicamente inducir el daño del cardiomiocito y muerte celular, fenómeno llamado daño por isquemia-reperfusión (I-R) (2,3). Por esta razón son necesarias intervenciones farmacológicas para reducir mejor el tamaño del infarto, preservar la función del ventrículo izquierdo, mitigar el remodelado y, por consiguiente, mejorar la supervivencia en pacientes con IAM.
La isquemia miocárdica induce fenómenos de muerte celular por necrosis y apoptosis, que son los causantes del infarto final. Durante el periodo isquémico se produce necrosis pero además se activa la apoptosis. La apoptosis es un proceso que requiere energía (es un proceso dependiente de oxígeno y ATP), por lo que -aunque los genes proapoptóticos se activen durante la isquemia- el programa apoptótico no se lleva a cabo hasta la reperfusión (cuando el oxígeno está nuevamente disponible para sintetizar ATP). Es decir, la apoptosis se activa durante la isquemia pero se efectúa durante la reperfusión. Por ello, la reperfusión, aunque beneficiosa para evitar la muerte por necrosis, paradójicamente puede tener efectos perjudiciales al poner en marcha la muerte celular por apoptosis. Este es el origen de la lesión por isquemia reperfusión (I-R), que es responsable hasta del 50% del tamaño final del IAM (2,3). Por ello, se postula que fármacos antiapoptóticos podrían disminuir la lesión por I-R.
La esfingosina-1-fosfato (S1P) ha recibido recientemente mucha atención por sus efectos antiapoptóticos y citoprotectores y su potencial para mejorar el daño por I-R (4). S1P es un lipofosfolípido bioactivo derivado del lípido de membrana esfingomielina (5). De una manera importante, se ha demostrado recientemente que S1P reduce el daño por I-R en el hígado (6), riñón (7), páncreas (8), y en el cerebro (9). En experimentos in vitro, S1P aumenta la supervivencia del cardiomiocito en condiciones de hipoxia (10,11), y reduce el tamaño del infarto en experimentos ex vivo de preparaciones de corazones aislados (12,13). Además, experimentos in vivo iniciales también apoyan el papel cardioprotector de S1P en ratones durante el IAM (14,15).
Nuestra estrategia final consiste en aplicar un fármaco antiapoptótico al paciente que sufre un IAM ya durante el traslado en ambulancia del paciente al hospital, es decir antes de la reperfusión. Para facilitar la aplicación de nuestros hallazgos a la práctica clínica, elegimos un agonista de los receptores de S1P (S1P-R). Fingolimod, un derivado sintético de la miriocina (un metabolito del hongo Isariasinclairii), es el único agonista de S1P-R disponible para el uso clínico en humanos (16). Fingolimod reduce las recurrencias y las lesiones en cerebro (medidas por resonancia magnética) en pacientes con esclerosis múltiple recurrente-remitente (17,18), y fue aprobado por la Asociación Americana de Medicamentos y Alimentos (FDA) en Septiembre de 2010 como tratamiento de primera línea para estos pacientes. Además, la activación de S1P-R mediada por Fingolimod podría ser una terapia prometedora con el objetivo de mejorar la recuperación del miocardio durante el IAM. De hecho, estudios preliminares han demostrado que Fingolimod reduce la apoptosis del cardiomiocito en experimentos in vitro en condiciones de hipoxia (4,19-21). Sin embargo, aunque estos estudios preliminares son muy prometedores, el efecto cardioprotector de Fingolimod no se han estudiado todavía en modelos in vivo.
Nuestra hipótesis consiste en que la activación de S1P-R con Fingolimod durante la isquemia reduce la apoptosis de los cardiomiocitos dando lugar a un aumento de la recuperación miocárdica, disminución del tamaño de infarto y reducción del remodelado del VI en un modelo porcino de I-R.
2. material y métodos
2.1. Diseño del estudio
Utilizamos cerdos Yorkshire albinos (20±2kg) para este estudio, que fueron alojados en un animalario acreditado por la Asociación para la Evaluación y Acreditación del Cuidado de Animales de Laboratorio. Todos los experimentos se realizaron manteniendo a los animales anestesiados. Justo antes de la inducción del IM se realizaron ecocardiografías 2D y 3D. Seguidamente se llevó a cabo un modelo de I-R tal y como está descrito previamente por nuestro grupo (22).
En el primer grupo de animales, indujimos IAM en 10 cerdos (5 tratados con Fingolimod 1mg/kg IV 15 minutos antes de la reperfusión, 5 controles), que fueron sacrificados a las 24 horas post-IAM para la valoración de la apoptosis y la activación de rutas moleculares en la zona del borde del miocardio isquémico.
Para el análisis a largo plazo, indujimos IAM en 14 animales. Los animales fueron aleatorizados a tratamiento con Fingolimod (1mg/kg IV, Cayman) 15 minutos antes de la reperfusión y una vez al día durante los siguientes 3 días, o con salino (grupo control) con un diseño 1:1. Una semana post-IAM se tomaron imágenes por resonancia magnética (RM, para la cuantificación de la función del VI, área en riesgo –AAR-23 y tamaño de IAM) y ecocardiografía 2D y 3D para estudiar la mecánica del VI. Así mismo se repitieron estas técnicas 1 mes después del IM. Al finalizar las técnicas de imagen los animales fueron evaluados por técnicas invasivas de hemodinámica (análisis de presión-volumen) y seguidamente sacrificados para la toma de muestras de tejido.
2.2. Inducción del Infarto Agudo de Miocardio
Doce horas antes de la inducción del IM todos los animales recibieron una dosis oral de clopidogrel150 mg. El día del IM la anestesia de los animales se indujo con telazol (6 mg/kg i.m.) y se mediante administración intravenosa constante de propofol (10 mg/kg/hr). Los cerdos fueron medicados con 3000 UI de heparina IV, 75 mg de amiodarona IV y atropina (0,04mg/kg). Se administró una perfusión continua a 2 ml/kg/hr que contenía amiodarona 75 mg, cloruro de potasio 10mEq y atropina 0,04 mg/kg.
Llevamos a cabo el acceso arterial a través de la arteria femoral por punción percutánea. Colocamos un introductor arterial de 7,5 French mediante técnica de Seldinger e insertamos un catéter guía coronario de 7 French desplazándolo hasta la arteria aorta ascendente hasta alcanzar el tronco coronario izquierdo. Realizamos una angiografía en el momento inicial. Bajo fluoroscopia, implantamos una guía coronaria en la arteria coronaria descendente anterior (DA). Sobre esa guía coronaria, implantamos un catéter balón de angioplastia (TREK OTW Coronary Dilation Catheter 4.0 x 15 mm, Abbot Vascular) en el segmento proximal de la DA inmediatamente proximal a la salida de la primera rama septal. De esta manera indujimos el IM por oclusión de la DA proximal durante 60 minutos inflando el globo en la arteria coronaria a 10 atm. A lo largo de todo el proceso, mantuvimos a los animales monitorizados con un EKG y un sistema invasivo para la medida de la presión sanguínea. Fue necesario prestar una atención especial para la detección temprana de arritmias ventriculares para llevar a cabo la desfibrilación. Tras 60 minutos, deshinchamos el balón y retiramos el catéter de la DA. La reperfusión fue documentada con un angiograma final. Retiramos entonces el introductor arterial y realizamos compresión manual sobre la zona de punción durante unos 15-20 minutos para hemostasia. El cuidado tras este procedimiento consistió en cefazolina (25 mg/kg) como profilaxis antibiótica, buprenorfina como profilaxis analgésica (0,1 mg/kg), e infusión continua de amiodarona (150 mg en 500 ml de salino, a pasar en 12 horas) como profilaxis antiarrítmica.
2.3. Eutanasia
Inmediatamente tras la realización de RM y ecocardio, los animales fueron sacrificados por inyección intravenosa de cloruro de potasio (20 mEq inyectados a través del acceso venoso central en la vena femoral), se extrajeron los corazones y se realizaron siete cortes del mismo grosor a lo largo del eje corto del VI. Se recogieron muestras de tejido de la zona de la cicatriz, borde y remota, parte se incluyó y congeló en medio OCT (TissueTek, California) para los estudios histológicos y parte se congeló para el análisis molecular. Visualizamos el tamaño final del infarto en los cortes restantes a través de la tinción con cloruro de 2,3,5-trifenil tetrazolio (TTC) y se tomaron imágenes de alta resolución pudiendo diferenciar el área del infarto (blanco) y el miocardio normal (rojo). Las imágenes se cuantificaron utilizando el software Image J (National Institutes of Health, Bethesda, MD). El volumen de infarto se expresó como porcentaje del volumen total del miocardio.
2.4. Imagen cardiaca por resonancia magnética
Los estudios por resonancia magnética (RM) se realizaron en un equipo de RM de 3.0 Tesla (Achieva, Philips Medical System, Netherlands). Se realizó una secuencia de cine en el eje corto del VI (TR 3,6 ms, TE 1,6 ms, flipangle 45º, campo de visión 250 x 250 mm)desde la base hasta el ápex del VI para la cuantificación de los volúmenes, masa y fracción de eyección del VI (FEVI). A continuación realizamos una secuencia spin echo potenciada en T2(STIR, TR 2-3 latidos, TE 60 ms, TI 210 ms, campo de visión 250 x 250 mm, SENSE factor 3) para la medida del edema y determinación del área en riesgo (AAR). Realizamos realce tardío de gadolinio 15 minutos después de la administración del gadolinio (Magnevist, 0,2 mmol/kg) usando una secuencia de inversión-recuperación (TR 9 ms, TE 5,4 ms, TI optimizado para anular el miocardio sano, campo de visión 250 x 250 mm). Después del realce tardío, infundimos dobutamina a 5µg/kg/min y repetimos la secuencia SSFP para la determinación de la reserva contráctil.
Todas las imágenes de RM fueron analizadas de manera ciega usando un software comercial (Extended MR Workspace, Philips Medical Systems, Netherlands). Se trazaron los contornos del epicardio y del endocardio en cada una de las imágenes SSFP para la obtención de los volúmenes sistólicos y diastólicos finales (LVEDV y LVESV), FEVI y la masa del VI; por convención, los músculos papilares se incluyeron en la cavidad del VI. Para la determinación del AAR y de tamaño de IAM se delimitaron manualmente los bordes epicárdicos y endocárdicos de cada uno de los cortes del eje corto en las secuencias STIR e inversión-recuperación, respectivamente; las áreas hiperintensas (intensidad de señal superior a tres veces la desviación estándar del miocardio remoto sano) definió el AAR y la escara. El VI se dividió en 16 segmentos y a cada segmento se le asignó una puntuación del cero al cuatro, teniendo en cuenta el porcentaje de miocardio cicatrizado (0- sin cicatriz, 1- menos del 25% de cicatriz, 2- del 25 al 50% de cicatriz, 3- del 50 al 75% de cicatriz, 4- más del 75% de cicatriz). La puntuación final (el índice de transmuralidad de la cicatriz) fue calculado como la suma de los 16 segmentos. El miocardio preservado se calculó como la diferencia entre el área en riesgo (edema medido en secuencias SE potenciadas en T2) y el tamaño de la cicatriz (medida a partir del realce tardío de gadolinio). El índice de miocardio preservado del miocardio se calculó como [Área en Riesgo (g) – Tamaño de IAM (g)] / Área en Riesgo (g) y expresado como porcentaje del VI.
2.5. Ecocardiografía
Realizamos ecocardiografía 2D con un sistema iE33 con transductor S5 (Philips Medical Systems, Andover, MA) de acuerdo con las recomendaciones de la Sociedad Americana de Ecocardiografía y de la Sociedad Europea de Ecocardiografía (24,25). Las imágenes fueron analizadas con el software comercial Q-Lab (Philips Medical Systems, Andover, MA).Las imágenes ecocardiográficas en 3D del volumen total se adquirieron con este mismo sistema iE33 utilizando un transductor X3 (Philips Medical Systems, Andover, MA). Se tuvo un especial cuidado a la hora de incluir la cavidad del VI dentro del volumen piramidal del transductor. Los ajustes para la adquisición de la imagen fueron optimizados para una visualización óptima del endocardio y al menos se adquirieron y guardaron tres datos para las posteriores medidas. La imagen en 3D de la superficie del endocardio se construyó de una forma semiautomática a lo largo del ciclo cardiaco. Seguidamente se ajustó manualmente la superficie si era necesario. Para determinar la deformación global del miocardio, realizamos un análisis de strain 3D utilizando el software 4D (Tom Tec Imaging Systems, Germany). Se seleccionaron vistas apicales a diferentes niveles de dos-cámaras, cuatro-cámaras y eje corto al final de la diástole. El VI se dividió en 16 segmentos de tres dimensiones usando una segmentación estándar. Se midieron los strains longitudinal (LS), circunferencial (RS) y radial (RS) globales en 3D y se hizo la media de los 16 segmentos. El índice de esfericidad del VI se calculó como el cociente eje mayor (longitudinal) del VI / eje menor (transversal) del VI en un eje apical de cuatro-cámaras (Figura 5C), como estaba descrito previamente (26). Calculamos el grosor relativo de la pared como 2 x Grosor de la Pared Posterior / Diámetro del VI al final de la diástole (26).
2.6. Hemodinámica invasiva con microcatéter
Las curvas de presión-volumen del VI se obtuvieron un mes después del infarto e inmediatamente antes del sacrificio para determinar las diferencias hemodinámicas entre grupos. A través de la arteria femoral, introdujimos un microcatéter de tipo Millar de 7F (Millar Instruments Inc., Houston, TX) hasta el ápex del VI y tomamos durante las siguientes medidas: presión sistólica, presión al final de la diástole, tasa máxima de cambio de la presión en el VI (dP/dt) máx, y trabajo cardiaco; el índice de contractilidad fue calculado como (dP/dt) máx / (presión telesistólica de VI – presión telediastólica de VI). Cada parámetro se calculó a partir de la media de, al menos, tres ciclos cardiacos consecutivos.
2.7. Niveles plasmáticos de metanefrinas
Se tomaron muestras de plasma quince minutos tras la inserción del introductor arterial y teniendo al animal anestesiado y en un estado de reposo. Los niveles de metanefrinas se determinaron utilizando un método de ELISA (Rocky Mountain Diagnostics Colorado Springs, Co).
2.8. Análisis histológico
Los corazones incluidos en OCT se cortaron a 8 µm y fueron usados para analizar fibrosis y tamaño de los cardiomiocitos en el tejido miocárdico remoto, no isquémico y para la determinación de la apoptosis en el tejido del borde del infarto. Para analizar el tamaño celular, se realizó una inmunohistoquímica para vinculina (#V9131, Sigma-Aldrich). Para ello utilizamos tres cortes de cada muestra que fueron permeabillizados y bloqueados con Triton 0,3% y con BSA 1% respectivamente. El anticuerpo primario anti-vinculina a una dilución 1:100 fue incubado durante toda la noche y a 4ºC. Tras varios lavados con PBS, los cortes fueron incubados con el anticuerpo secundario, Alexa Fluor 488 Goat Anti-mouse (#A11001, Invitrogen) a una dilución 1:500 durante 1h a temperatura ambiente. Tras varios lavados con PBS, las preparaciones se montaron con medio de montaje con DAPI (#H-1500, Vector Lab) y se tomaron imágenes digitales (microscopio Zeiss Axioplan2 y el programa Zeiss Axio Vision, Micro-optik). El área y tamaño de los cardiomiocitos fueron cuantificados con el programa Image J (National Institutes of Health, Bethesda, MD).
Para la determinación de la fibrosis intersticial, se tiñeron 3 cortes de cada muestra con Rojo Sirio (Spectrum Chemical) de acuerdo con las especificaciones de la casa comercial. Las imágenes fueron realizadas con luz polarizada (microscopio Zeiss Axioplan2 y el programa Zeiss Axio Vision (Micro-optik)). Se cuantificó y analizó el área del miocardio positiva para esta tinción con el programa Image Pro Plus.
Para la determinación de la apoptosis celular vía fragmentación del DNA se utilizó un kit ensayo comercial de TUNEL (ApoTag® Florescein In Situ Apoptosis Detection Kit, Millipore) siguiendo el protocolo del manual. Para las cuantificaciones se analizó un mínimo de 10 campos con un objetivo 20X por corte.
2.9. Expresión de proteínas
Para la preparación del extracto proteico de las muestras de corazón se homogeneizaron unos 50 mg de miocardio congelado de cada uno de los animales (RIPA buffer (#R0278, Sigma-Aldrich) con inhibidores de proteasas y un sistema específico de homogeneizado (Fast Prephomogenizer). Después de 20 minutos de centrifugación a 30.000 g, la parte insoluble de la muestra fue descartada y el sobrenadante almacenado a -80ºC para posteriores estudios bioquímicos. Para el estudio de la expresión de proteínas, se cargaron 50 µg de proteína junto con tampón de carga (40% β-mercaptoetanol, 8% SDS, 40% glicerol, 0,025% azul de bromofenol, and 0,25 mmol/L Tris, pH 6,4), y se llevó a cabo la separación de las proteínas por electroforesis en geles de acrilamida al 12%. Una vez separadas las proteínas, se transfirieron a una membrana de nitrocelulosa y se incubó durante toda la noche y a 4ºC con los anticuerpos específicos contra las proteínas que queríamos estudiar (Cell Signaling), anti-phospho-p44/42 MAPK (Erk1/2), p44/42 MAPK (Erk1/2), phospo-Akt (Thr308), and Akt (Thr308). La dilución usada para todos ellos fue 1:1000. Tras varios lavados con TBST, se las membranas se incubaron con el anticuerpo secundario conjugado con peroxidasa durante 1 hora a 4ºC (#RPN4301, Amersham) a una dilución. Un anticuerpo monoclonal anti-GAPDH (#G8795, Sigma-Aldrich) se usó como control de carga. Para la visualización de las bandas en las películas autorradiográficas utilizamos el sustrato quimioluminiscente ECL-Plus (#RPN2132, Amersham). Las películas fueron densitometradas y analizadas con el programa Image J.
2.10. Análisis estadístico
Los resultados están expresados como la media ± error estándar. La comparación entre ambos grupos, cerdos tratados con Fingolimod y grupo control, se realizó con el test t-test para las medidas tomadas en un único tiempo. Para aquellas medidas tomadas a una semana y a un mes después de la inducción del IM, los datos se analizaron mediante una ANOVA de medidas repetidas seguidos de un análisis post-hoc Tukey para determinar si había diferencias entre los grupos. Todos los análisis se realizaron con el programa estadístico SPSS 18.0 (SPSS Inc., Chicago, Illinois). Únicamente fueron consideradas significativas aquellas con una p < 0,05.
3. resultados y discusión
La activación de S1P-R con Fingolimod en el periodo pre-reperfusión redujo la apoptosis de los cardiomiocitos en la zona isquémica 24 horas después del IAM
La activación de S1P con Fingolimod dio lugar a una reducción significativa del número de núcleos positivos para la tinción de TUNEL indicando una disminución de la apoptosis de los cardiomiocitos (Figura 1A-B) en el grupo Fingolimod. La técnica de Western blot confirma estos resultados pues se observa una disminución de la activación de proteínas proapoptóticas Caspasa-3 y p53 y un aumento en la activación de la proteína antiapoptótica Bcl-2.
Además, evaluamos la ruta molecular RISK (Reperfusion Injury Salvaje Kinases), la principal ruta de protección de los cardiomiocitos contra el daño por I-R, que actúa a través de la fosforilación de Akt, PKC-ε y GSK-3β. Nuestros resultados muestran que la fosforilación de Akt, PKC-ε y GSK-3β estaba incrementada significativamente en los cerdos tratados con Fingolimod comparado con los controles (Figura 1C-D). Estos datos indican que la activación de S1P-R con Fingolimod en el periodo pre-reperfusión activa la ruta RISK y promueve la citoprotección de los cardiomiocitos.
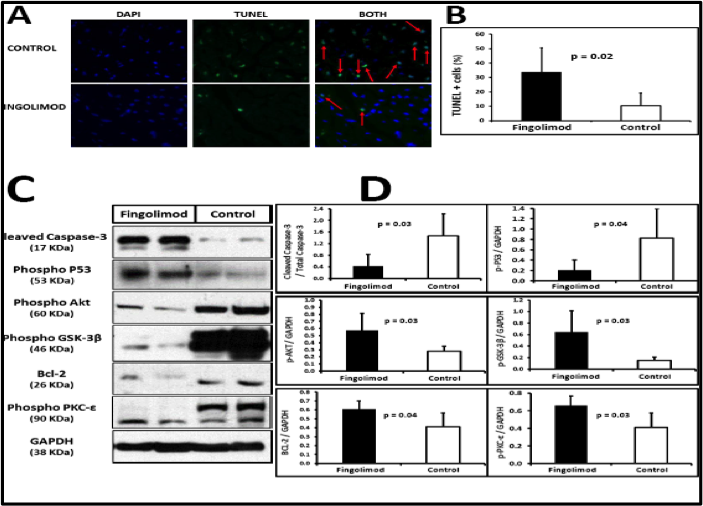
Figura 1.-El tratamiento con Fingolimod redujo la apoptosis en el miocardio isquémico. A: Imágenes representativasde DAPI (indicando los núcleos en azul) y de TUNEL (indicando los núcleos apoptóticos en verde) en inmunofluorescencia. Los núcleos positivos para TUNEL se calcularon a partir de las imágenes superpuestas TUNEL+DAPI. B: La cuantificación de las células TUNEL+ muestra menor porcentaje de células apoptóticas en los cerdos tratados Fingolimod. C y D: Western blot revela menor activación de proteínas pro-apoptóticas(caspasa-3 activa o cleaved caspase-3, p-P53) y mayor activacíon de proteínas anti-apoptóticas(p-Akt, p-GSK3-β, Bcl-2, p-PKC-ε) en el miocardio isquémico de los cerdos tratados con Fingolimod. Imágenes representativas de Western bloten C, cuantificación en D.
La activación de S1P-R con Fingolimod redujo el tamaño de infarto, aumentó el miocardio preservado y mejoró la función sistólica del VI (FSVI) 1 semana después del IAM
La activación de S1P-R con Fingolimod dio lugar a un tamaño de infarto significativamente menor tanto en términos de masa infartada absoluta como en porcentaje de miocardio de VI infartado en comparación con los cerdos control (Figura 2A-B, Tabla 1). Dado que AAR medida mediante RM fue similar en ambos grupos (Figura 2C-D, Tabla 1), la activación de S1P-R en el periodo pre-reperfusión aumentó el porcentaje de miocardio preservado (Figura 2E, Tabla 1). De hecho, el porcentaje de miocardio preservado fue 5 veces mayor en los cerdos tratados con Fingolimod que en el grupo control. El índice de transmuralidad, una medida alternativa por RM del tamaño de infarto, también era significativamente menor al cabo de 1 mes en los cerdos tratados con Fingolimod (Figura 2F).
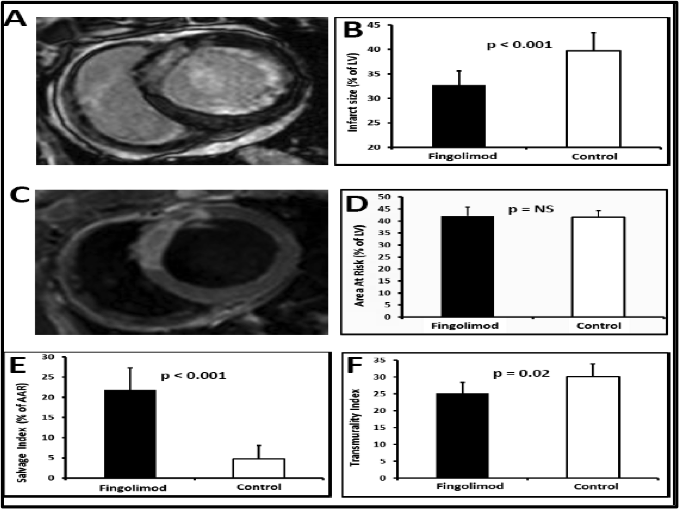
Figura 2.- El tratamiento con Fingolimodredujo el tamaño de infarto y aumentó el miocardio preservado una semana post-IAM. A: Imágenes representativas de RM cardiaca para realce tardío de gadolino. Las zonas hiperintensas (blanco) representan el miocardio infartado. B: Cuantificación de tamaño mediante RM; el tamaño de IAM era menor en los cerdos tratados con Fingolimod. C: Imágenes representativas de RM cardiaca para edema. Las zonas hiperintensas (blanco) representan el área en riesgo (AAR), el miocardio sometido a isquemia. D: Cuantificación de AAR mediante RM, que era similar en ambos grupos. E: El tratamiento con Fingolimodaumentó significativamente el miocardio preservado. F: El índice de transmuralidad, una medida alternative de tamaño de IAM, asisismo mostró que el tamaño de IAM era significativamente menor en los cerdos tratados con Fingolimod. Infarctsize: Tamaño de IAM. Area At Risk: Miocardio en riesgo. MyocardialSalvage: Miocardio preservado. TransmuralityIndex: Índice de transmuralidad.
Inicialmente, FEVI por ecocardiografía-3D y los strain tridimensionales eran similares en ambos grupos pre-IAM (Tabla 2),por lo que había diferencias en la función sistólica del VI antes de la inducción del infarto. Queremos resaltar que el aumento en miocardio preservado debido a la activación de S1P-R con Fingolimod mejoró la función contráctil al cabo de 1 semana. La FEVI era significativamente mayor en el grupo Fingolimod que en el grupo control tanto por RM (Figura 3A, Tabla 1) como por ecocardiografía-3D (Tabla 2). Además, 1 semana post-IAM, tanto el strain tridimensional longitudinal (3D-LS), el strain tridimensional radial (3D-RS) como el strain tridimensional circunferencial (3D-CS) estaban más preservados en los cerdos tratados con Fingolimod que en los controles (Figura 4, Tabla 2).
La activación de S1P-R redujo el tamaño de IAM y mejoró la FSVI un mes post-IAM
Al igual que 1 semana post-IAM, el tamaño de infarto medido por RM un mes post-IAM era significativamente menor en los cerdos tratados con Fingolimod en comparación con los controles (Tabla 1). Además, el índice de transmuralidad también era significativamente menor en los cerdos tratados con Fingolimod. La reducción en el tamaño final de infarto fue confirmada por histología utilizando la tinción con TTC (23,32±3,78 vs 29,73±4,92 % del volumen del VI para el grupo Fingolimod y control respectivamente, p=0,02). Hubo además una excelente correlación entre el tamaño de infarto al cabo de un mes cuantificado por LGE y por tinción con TTC (r=0,90, p=0,01).
Tabla 1.- Resultados de RM cardiaca una semana y un mes post-IAM.
1 semana post-IAM |
1 mes post-IAM |
|||||
Control |
Fingolimod |
p-Value |
Control |
Fingolimod |
p-Value |
|
Tamaño de IAM (% de VI) |
39,69±3,71 |
32,76±2,88 |
0,002 |
32,22±4,44 |
25,47±4,60 |
0,01 |
Área en Riesgo (% de VI) |
41,63±2,74 |
42,01±3,74 |
0,83 |
|||
Miocardio preservado (% de Área en Riesgo) |
4,78±3,28 |
22,86±5,39 |
<0,001 |
|||
Miocardio preservado (% de VI) |
1,93±1,22 |
9,26±2,84 |
<0,001 |
|||
Índice de transmuralidad |
26,43±3,64 |
20,83±3,87 |
26,43±3,64 |
20,83±3,87 |
0,04 |
|
Masa de VI (g) |
60,54±4,71 |
52,43±2,43 |
0,01 |
72,18±5,53 |
62,97±3,91 |
0,005 |
VTDVI (ml) |
83,70±15,35 |
73,57±15,87 |
0,25 |
118,33±22,14 |
96,96±15,98 |
0,08 |
VTSVI (ml) |
55,86±12,08 |
44,72±11,14 |
0,1 |
79,61±19,15 |
55,09±13,50 |
0,02 |
FEVI (%) |
33,61±2,84 |
39,61±4,16 |
0,008 |
33,30±4,26 |
43,68±5,85 |
0,001 |
Volumen Latido de VI (ml) |
27,86±3,56 |
29,01±5,35 |
0,64 |
38,73±3,77 |
41,85±5,14 |
0,23 |
Peak ejection rate (ml/ms) |
0,16±0,04 |
0,29±0,04 |
<0,001 |
|||
ΔLVEF, Incremento de FEVI desde 1 semana a 1 mes post-IAM (%) |
0,24±2,20 |
3,93±1,83 |
0,004 |
|||
Reserva contráctil (FEVI maxima con dobutamina, %) |
35,94±8,21 |
55,66±10,34 |
0,007 |
|||
Reserva contráctil (ΔLVEF con dobutamine, %) |
2,88±3,70 |
11,99±5,56 |
0,01 |
|||
FEVI: Fracción de Eyección del VI. VTDVI: Volumen TeleDiastólico del VI. VTSVI: Volumen TeleSistólico del VI.
La activación de S1P-R también resultó en una mejora de la función sistólica un mes post-IAM. Los animales tratados con Fingolimod mostraron mayor FEVI que los cerdos control tanto por RM (Tabla 1) como por ecocardiografía-3D (Tabla 2). Además, en los cerdos Fingolimod la FEVI por RM mejoró en el periodo comprendido entre una semana y un mes mientras que se mantuvo igual en los cerdos control (Figura 3B, cambio absoluto de FEVI 3,9±1,8% vs. -0,2±2,2% en el grupo Fingolimod y control respectivamente, p=0,004). Además, la reserva contráctil con dobutamina estaba aumentada en los animales tratados con Fingolimod (Figura 3C), incremento absoluto en la FEVI 12±5,5% vs. 2,8±3,7% en el grupo Fingolimod y control respectivamente, p=0,01).
Tabla 2.- Resultados de ecocardiografía 3D pre-IAM, una semana y un mes post-IAM.
Control |
Fingolimod |
P-value |
|
3D-FEVI pre-IAM (%) |
71,59 ± 3,03 |
71,50 ± 1,34 |
0,95 |
3D-FEVI 1 semana post-IAM (%) |
32,98 ± 4,88 |
40,15 ± 5,02 |
0,04 |
3D-FEVI 1 mes post-IAM (%) |
34,24 ± 5,26 |
44,71 ± 6,96 |
0,001 |
3D-VTDVI pre-IAM (mL) |
43,81 ± 4,38 |
40,97 ± 6,46 |
0,37 |
3D-VTDVI 1 semana post-IAM (mL) |
66,94 ± 6,40 |
62,32 ± 7,68 |
0,31 |
3D-VTDVI 1 mes post-IAM (mL) |
93,99 ± 17,70 |
81,03 ± 15,39 |
0,18 |
3D-VTSVI pre-IAM (mL) |
12,4 ± 1,15 |
11,67 ± 1,89 |
0,41 |
3D-VTSVI 1 semana post-IAM (mL) |
45,06 ± 7,25 |
37,30 ± 5,29 |
0,07 |
3D-VTSVI 1 mes post-IAM (mL) |
65,27 ± 17,35 |
45,33 ± 12,64 |
0,03 |
3D-Strain Longitudinal - pre-IAM (%) |
-21,4 ± 1,45 |
-22,13 ± 3,0 |
0,60 |
3D-Strain Longitudinal - 1 semana post-IAM (%) |
-9,33 ± 2,81 |
-14,43 ± 1,77 |
0,005 |
3D-Strain Longitudinal - 1 mes post-IAM (%) |
-11,35 ± 1,70 |
-16,34 ± 1,95 |
<0,001 |
3D-Strain Circunferencial- pre-IAM (%) |
-27,18 ± 4,08 |
-24,5 ± 5,3 |
0,37 |
3D-Strain Circunferencial – 1 semana post-IAM (%) |
-10,73 ± 2,72 |
-17,06 ± 5,35 |
0,04 |
3D-Strain Circunferencial – 1 mes post-IAM (%) |
-11,86 ± 3,81 |
-16,6 ± 3,94 |
0,03 |
3D-Strain Radial- pre-IAM (%) |
129,42 ± 46,31 |
120,21 ± 31,41 |
0,69 |
3D-Strain Radial – 1 semana post-IAM (%) |
26,94 ± 9,82 |
55,95 ± 20,42 |
0,03 |
3D-Strain Radial – 1 mes post-IAM (%) |
32,77 ± 10,59 |
62,51 ± 20,45 |
0,005 |
FEVI: Fracción de Eyección del VI. VTDVI: Volumen TeleDiastólico del VI. VTSVI: Volumen TeleSistólico del VI.
Las técnicas de hemodinámica invasiva corroboraron la mejor función sistólica en el grupo Fingolimod, pues presentaba dP/dtmáx, trabajo latido (strokework) índice de contractilidad y presión telesistólica del VI significativamente mayores que los controles (Figura 3D-F, Tabla 3).
Para caracterizar mejor la FSVI, determinamos la función mecánica del VI mediantes strain-3D. Los cerdos tratados con Fingolimod mostraron significativamente mejores 3D-LS, 3D-CS y 3D-RS un mes post-IAM que los controles (Tabla 2 y Figura 4).
La activación de S1P-R con Fingolimod en el periodo pre-reperfusión mitigó el remodelado anatómico del ventrículo izquierdo un mes después del IM.
Las características anatómicas del remodelado psot-IAM del VI son dilatación del VI con incremento en los volúmenes del VI, hipertrofia compensatoria del VI con aumento en la masa del VI y cambios en la morfología del VI (desde la geometría elíptica habitual hacia una conformación más esférica). La excesiva activación de la respuesta neurohormonal con activación del sistema simpático (expresado como altos niveles de catecolaminas en plasma) también es un componente importante.
Tabla 3.- Medidas de funcion sistólica de VI mediante hemodinámica invasiva un mes post-IAM.
Control |
Fingolimod |
P-value |
|
Presión TeleSistólica de VI (mmHg) |
95,00 ± 10,88 |
116,60 ± 19,68 |
0,03 |
dP/dtmax (mmHg/s) |
1056,15 ± 261,03 |
1865,88 ± 147,46 |
< 0,001 |
Índice de Contractilidad (1/s) |
13,84 ± 1,53 |
19,06 ± 3,11 |
< 0,001 |
Trabajo Latido |
2395,67 ± 401,80 |
3269,91 ± 968,46 |
0,047 |
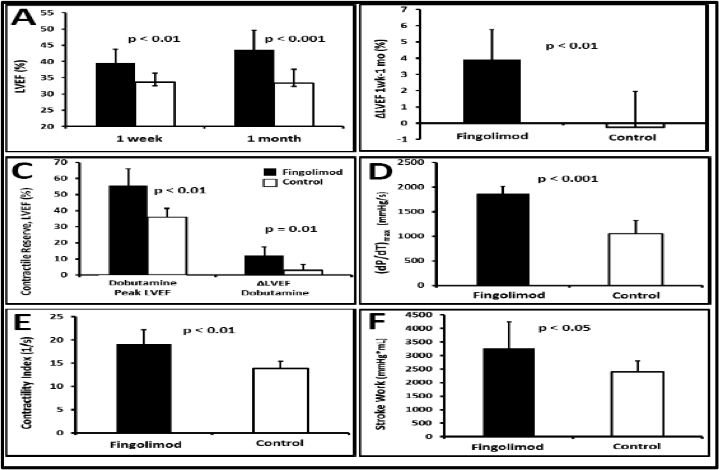
Figura 3.- El tratamiento con Fingolimod mejoró la funcion sistólica post-IAM del VI evaluada mediante RM cardiaca (A-C) y mediante hemodinámica invasiva (curvas de presión-volumen, D-F). Los cerdos tratados con Fingolimod (columnas negras) mostraron una mejor función sistólica del VI comparado con el grupo control (columnas blancas): A) FEVI 1 semana post-IAM y 1 mes post-IAM, B) mejoría de la FEVI desde 1 semana post-IAM a un mes post-IAM, C) reserva contráctil con dobutamina, D) (dP/dt)max, E) índice de contractilidad, and F) trabajo latido. LVEF: Fracción de Eyección del VI o FEVI. Contractile Reserve: Reserva Contráctil. ContractilityIndex: índice de contractilidad. SrokeWork: Trabajo Latido.
La RM mostró que los animales tratados con Fingolimod presentaban una masa del VI significativamente menos que los controles (Tabla 1). Cabe destacar que existía una correlación inversa entre la masa del VI y la FEVI (r=-0,72, p<0,001), y una correlación directa entre la masa del VI y el tamaño del infarto (r=0,65, p<0,001). Esto indica que los animales con mayor tamaño de infarto y menor FEVI muestran una mayor hipertrofia compensatoria. Los animales tratados con Fingolimod también presentaron menor índice de grosor de la pared (0,34±0,03 vs. 0,40±0,05, p=0,03), confirmando que el grupo Fingolimod presentaba una respuesta hipertrófica menor.
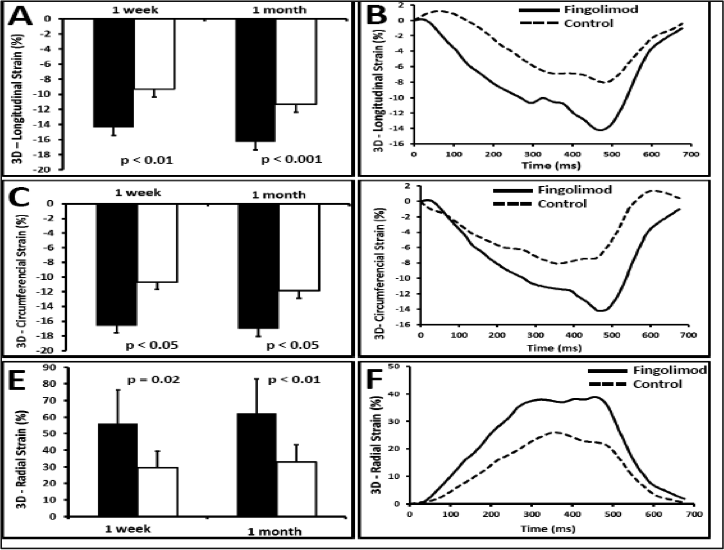
Figura 4.- El tratamiento con Fingolimod mejoró la mecánica miocárdica evaluada mediante strain por ecocardiografía tridimensional. Los cerdos tratados con Fingolimod (columnas negras) mostraron una mejor contractilidad miocárdica comparado con el grupo control (columnas blancas), tanto en strain 3D longitudinal (A), como en strain 3D circunferencial (C) como en strain 3D radial (E) una semana post-IAM y un mes post-IAM. Los paneles B, D y F muestra ejemplos representativos de la curvas de strain tridimensional de ambos grupos (control y Fingolimod).
Además, la RM demostró que la activación de S1P-R daba lugar a una reducción en la dilatación del VI al cabo de un mes del IM. Específicamente, los cerdos tratados con Fingolimod mostraron significativamente menor Volumen Tele Sistólico (LVESV) que los controles (Tabla 1). Además encontramos una fuerte tendencia (aunque no estadísticamente significativa) a un menor Volumen TeleDiastólico (LVEDV) en los cerdos tratados con Fingolimod (p=0,08, Figura 5B, Tabla 1).
Para evaluar los cambios geométricos del VI se midió también el índice de esfericidad del VI por ecocardiografía 2D. El índice de esfericidad del VI fue 1,61±0,11 en los animales tratados con Fingolimod en comparación con 1,37±0,14 en el grupo control (Figura 5C y 5D, p=0,01), que indica una menor deformación del VI en el grupo Fingolimod.
Los niveles de metanefrinas en plasma estaban reducidos en los cerdos tratados con Fingolimod (285,11±366,85 vs. 774,21±158,42 pg/mL, p=0,045), lo que indica una menor activación neurohormonal simpática en el grupo Fingolimod.
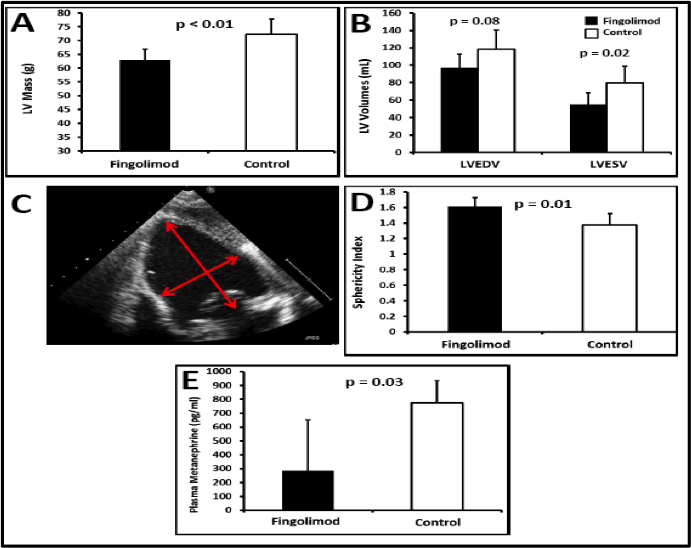
Figura 5.- El tratamiento con Fingolimod mitigó el remodelado anatómico post-IAM. A: Los animales tratados con Fingolimod mostraban menor masa de VI, lo que indica menor hipertrofia compensadora en el grupo Fingolimod. B: Los cerdos tratados con Fingolimod mostraban unos volúmenes telesistólicos (LVESV) menores que los controles, y una tendencia hacia unos menores volúmenes telediastólicos (LVEDV) que el grupo control. C: El índice de esfericidad del VI se calculó como el cociente eje mayor (longitudinal) del VI / eje menor (transversal) del VI en un eje apical de cuatro-cámaras en ecocardiografía bidimensional. D: Los animales tratados con Fingolimod mostraban menor cambio en el índice de esfericidad, lo que indica que sufrían menos cambios en la morfología, en la arquitectura, en la forma del VI. E: Los animales tratados con Fingolimod presentaban menores niveles plasmáticos de metanefrinas, lo que indica menor activación neurohormonal del sistema nervioso simpático en el grupo Fingolimod. LV Mass: Masa de VI. LV Volumes: Volúmenes de VI. LVEDV: Volumen telediastólico de VI. LVESV: Volumen telesistólico de VI. Sphericity index: índice de esfericidad.
El tratamiento con Fingolimod redujo la hipertrofia compensatoria del cardiomiocito y el acúmulo de fibrosis intersticial en el tejido miocárdico remoto, no infartado
Los cambios histológicos del remodelado post-IAM son principalmente hipertrofia celular de los cardiomiocitos y aumento de la fibrosis miocárdica intersticial en el tejido remoto no infartado.
El tamaño del cardiomiocito un mes post-IAM era menor en el grupo Fingolimod evaluado mediante marcajade vinculina por inmunohistoquímica. Los animales tratados con Fingolimod presentaron un perímetro celular menor (157,92±19,93 vs. 175,95±7,94 µm, p=0,045) y un área celular transversal menor (1751,56±436,39 vs. 2198,45±290,17 µm2 , p=0,049) que los controles (Figura 6A-E), lo cual indica una menor respuesta hipertrófica compensatoria en el grupo Fingolimod.
La hipertrofia del cardiomiocito en el contexto del remodelado del VI está mediada por la activación (fosforilación) de Akt y de ERK 1/2. En concordancia con los resultados histológicos, la activación de S1P-R por Fingolimod da lugar a una reducción significativa en la fosforilación de Akt (0,33±0,32 vs 0,94±0,5, p=0,02) and ERK1/2 (0,16±0,03 vs 0,82±0,08, p=0,04) (Figura 7).
Finalmente, existía menos fibrosis intersticial un mes post-IAM en el miocardio remoto no infartado en los animales tratados con Fingolimod en comparación con los controles (rojo picrosirio: 8,42±0,87% vs. 10,10±1,58% del miocardio respectivamente, p=0,03, Figura 6F-J).

Figura 6.- El tratamiento con Fingolimodredujo el remodelado histológico en el miocardio remoto no infartado un mes post-IAM. A-E: Medida del tamaño de los cardiomiocitos. Imágenes representativas de inmunohistoquímica con vinculina y DAPI (la membrana de los cardiomiocitosestá marcada en verde, los núcleos en azul) que muestra el tamaño de los cadiomiocitos en el grupo Fingolimod (A, detalle en B) y grupo control (C, detail in D). E: Los animales tratados con Fingolimod mostraban menor tamaño de cardiomiocitos que el grupo control, resultado de una menor hipertrofia de VI. F-J: Medida de fibrosis interstiticial miocárdica. Imágenes representativas de tinción con rojo sirio en brightfield (F y H, colágeno en rojo, cardiomiocitos en amarillo)y luz polarizada (G e I, colágeno en amarillo y verde, el resto del tejido en negro) de un cerdo del grupo Fingolimod(F y G) y de un cerdo del grupo control (H e I). J: Los animales tratados con Fingolimod mostraban menor porcentaje de fibrosis intersticial en miocardio que los controles. Cardiomyocyte perimeter: Perímetro de los cardiomiocitos.
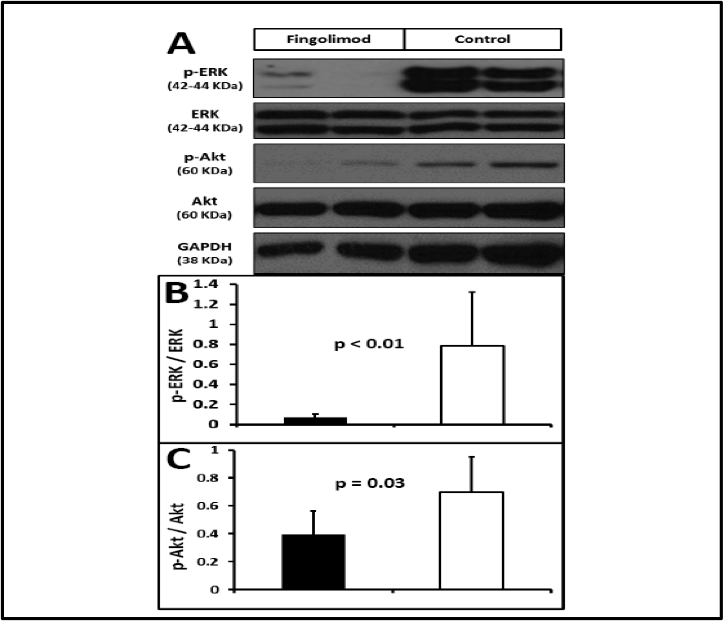
Figura 7.- El tratamiento con Fingolimod redujo el remodelado molecular un mes post-IAM, pues había menor activación (fosforilación) de ERK1/2 y de Akten el miocardio remoto no infartado un mes post-IAM. A: Western blotde p-ERK, ERK, p-Akt, Akt, y GAPDH (control de carga). B: Los animales tratados con Fingolimod presentaban menor activación de ERK1/2, demostrado por un menor cociente pERK/ERK. Este hallazgo apoya una menor hipertrofia en el grupo Fingolimod. C: El tratamiento con Fingolimod se asociaba con una menor activación (fosforilación) de Akt, consistente con menor estímulo hacia la hipertrofia de VI. p-ERK: ERK1/2fosforilado. P-Akt: Aktfosforilado.
4. DISCUSIÓN
En este estudio examinamos los efectos cardioprotectores del agonista del receptor de S1P Fingolimod en un modelo porcino de I-R. El principal hallazgo de este estudio es que la activación de S1P-R con Fingolimod inmediatamente pre-reperfusión da lugar a un mayor miocardio preservado, una disminución del tamaño de infarto y una mejora en la función sistólica del VI. Estos efectos están mediados por la activación de la ruta molecular cardioprotectora RISK que promueve una reducción en la apoptosis en los cardiomiocitos. Además, el tratamiento pre-reperfusión con Fingolimod reduce el remodelado adverso del ventrículo izquierdo que tiene lugar tras el infarto, tanto a nivel macroscópico como a nivel microscópico, y produce una normalización del eje catecolaminérgico.
Nuestra estrategia final consiste en intentar reducir el tamaño de IAM mediante la administración de un fármaco antiapoptótico durante el traslado en ambulancia del paciente con IAM al hospital, es decir incluso antes de la reperfusión. Para facilitar la aplicación de nuestros hallazgos a la práctica clínica, elegimos Fingolimod, el único agonista de S1P-R aprobado para uso humano.
Papel de S1P en el daño por I-R
Limitar el tamaño del infarto es de gran importancia puesto que se trata de un parámetro clave en la morbilidad y mortalidad. La restauración del flujo sanguíneo en el miocardio isquémico puede paradójicamente activar apoptosis (daño por I-R), siendo la lesión por I-R responsable hasta del 50% del tamaño de IAM (2,3). El lisofosfolípido SP1 ha sido reconocido recientemente como un mediador clave en la fisiología celular (5). La esfingosina es un derivado del lípido de membrana esfingomielina por la vía de las ceramidas. S1P es producto de la fosforilación de la esfingosina, reacción catalizada por una esfingosinakinasa. El concepto del reóstato de esfingosina fue introducido para expresar el balance entre ceramida, que es proapoptótica, y S1P, que activa rutas de supervivencia (27). Dado el papel fundamental dela apoptosis en el daño por I-R, la función de S1P como antiapoptótico representa una nueva diana farmacológica para disminuir este daño.
S1P promueve la supervivencia de los cardiomiocitos en experimentos in vitro llevados a cabo en condiciones de hipoxia, tanto en células neonatales (10) como en células adultas (11) de ventrículo de rata. S1P induce además resistencia al daño por I-R en experimentos in vivo en corazones aislados de ratones (12) y de rata (13) wild-type. Los ratones deficientes en esfingosinakinasa (la enzima responsable de la síntesis de S1P) mostraban un daño miocárdico por I-R más severo que los controles (28), mientras que la sobreexpresión de esfingosinakinasa (mediante transfección con adenovirus) protegía al miocardio del daño por I-R (14). Además, ratones deficientes en S1P-R mostraron un tamaño de infarto mayor que los ratones controles (15). Asimismo, S1P es un mediador clave en el precondicionamiento y en el postcondicionamiento, dos estrategias de cardioprotección establecidas. De hecho, el precondicionamiento y el postcondicionamiento reducen el tamaño de infarto en corazones wild-type pero ninguno de ellos fue cardioprotector en corazones deficientes en esfingosinakinasa o en S1P-R (13,28,29). Finalmente, se ha demostrado que HDL reduce el tamaño de infarto in vivo (30); este efecto está mediado por S1P ya que el efecto cardioprotector de HDL se pierde en ratones deficientes en S1P-R y durante el tratamiento con antagonistas de S1P-R. Todos estos resultados apuntan hacia un prometedor papel de S1P en la prevención del daño por I-R. Nosotros decidimos estudiar Fingolimod en nuestros experimentos puesto que es el único agonista de S1P-R aprobado por la FDA, lo que facilitaría la aplicación de esta estrategia terapéutica a la práctica clínica.
La apertura del poro de permeabilidad mitocondrial (MPTP) es el responsable final de la apoptosis mediada por el daño por I-R. En concreto, está demostrado que la prevención de la apertura del MPTP reduce el tamaño del infarto (31). La principal ruta molecular involucrada en la inhibición de la apertura del MPTP es la ruta RISK (32), que conlleva la fosforilación (activación) de Akt y PKC-ε y la fosforilación (inactivación) de GSK3-β. De hecho, nuestros resultados demuestran que Fingolimod da lugar a la activación de Akt y PKC-ε y a la inactivación de GSK3-β a las 24 horas post-IAM. Estos datos son consistentes con estudios in vitro preliminares que muestran que Fingolimod previene la apoptosis de los cardiomiocitos expuestos a condiciones de hipoxia mediante la activación de la ruta RISK (19), demostrando nuestros resultados el mismo proceso in vivo. Como resultado, los cerdos tratados con Fingolimod presentaron una reducción de la apoptosis celular medida por ensayo TUNEL y un incremento de la expresión de la proteína antiapoptótica Bcl-2 24 horas después del IAM. Estos mecanismos celulares serían la causa de la mejora en la recuperación del miocardio y a la disminución del tamaño del infarto en los cerdos tratados con Fingolimod.
La activación de S1P-R con Fingolimod reduce el tamaño de infarto y mejora la función sistólica del VI
Nuestros resultados demuestran que Fingolimod reduce el tamaño de infarto, determinado por técnicas no invasivas (RM) y por tinción post-mortem (TTC). Ambos métodos mostraron una reducción del 20% en el tamaño del infarto en el grupo tratado con Fingolimod. Este resultado tiene mucha importancia ya que el tamaño de infarto es un predictor independiente de pronóstico de enfermedad cardiovascular, incluso tras ajustar por FEVI (33-37).
Existe siempre una variación entre animales en el tamaño de infarto debido a las variaciones de la anatomía coronaria de cada animal. Para solucionar este problema, no sólo medimos el tamaño de infarto sino que también calculamos el área en riesgo (el miocardio sometido a isquemia que será necrosado si no se trata a tiempo) (22,23). El miocardio preservado (la proporción de miocardio en riesgo que es salvado con una intervención terapéutica, en nuestro caso el tratamiento con Fingolimod) es 5 veces mayor en los cerdos tratados con Fingolimod que en los cerdos control. Este hecho es de gran importancia ya que el miocardio preservado per se es un parámetro que predice de forma independiente eventos cardiovasculares adversos, incluso tras ajustar con el tamaño de infarto (38).
Además, la reducción del tamaño de IAM se correlaciona con una mejora en la FSVI. El grupo Fingolimod mostró una FEVI mayor a una semana y a un mes post-IAM (tanto por RM como por Eco-3D) y una mayor recuperación de la FEVI que el grupo control. Además el grupo tratado con Fingolimod presentó una mejor reserva contráctil que el grupo control. Estos resultados son importantes ya que tanto una FEVI más preservada (39) como la presencia de reserva contráctil (36,40) indican ambos mejor pronóstico cardiovascular.
Encontramos asimismo que la deformación miocárdica estaba más preservada en los cerdos tratados con Fingolimod. La FEVI es un parámetro clínicamente muy utilizado pero que presenta ciertos inconvenientes (dependiente de precarga, asunciones geométricas que no son ciertas ante alteraciones de contractilidad segmentarias post-IAM). Por ello, recientemente se ha desarrollado el análisis de strain mediante ecocardiografía, una herramienta nueva que permite una evaluación más detallada de la mecánica del miocardio (41) y que predice eventos clínicos mejor que la FEVI en pacientes con IAM previo (42,43). Nuestros datos indican claramente que el tratamiento con Fingolimod mejora los tres strains tridimensionales (strain longitudinal, circunferencial y radial) en comparación con el grupo control. Estos hallazgos apoyan la reducción en el tamaño de infarto y la mejora de la FSVI en el grupo Fingolimod asimismo mostrada por FEVI y por las curvas de presión-volumen.
La activación de S1P-R con Fingolimod reduce el remodelado del VI
A pesar de que el tamaño de infarto es un predictor importante de la mortalidad tras un IM, la disfunción sistólica crónica y el remodelado adverso del VI tras el IM son importantes determinantes de la morbilidad del paciente y de los eventos a largo plazo (44). Nuestros datos son los primeros en demostrar que el tratamiento agudo con Fingolimod no sólo reduce el daño por I-R y disminuye el tamaño de infarto a corto plazo sino que sus efectos beneficiosos se extienden al periodo crónico pues además mitiga el desarrollo de miocardiopatía isquémica y remodelado cardiaco adverso.
El remodelado cardiaco se define como los cambios en la expresión genética, molecular, celular e intersticial que se manifiestan clínicamente como cambios en el tamaño, forma y funcionalidad del corazón ante el daño cardiaco (45). Los cambios macroscópicos que caracterizan el remodelado del VI conllevan dilatación de los volúmenes del VI e incremento en la masa del VI que dan lugar al desarrollo de una hipertrofia compensatoria y cambios en la arquitectura del VI haciendo que pierda su forma elíptica habitual (forma de bala) para tomar una forma más esférica. En nuestro estudio, los corazones de los cerdos tratados con Fingolimod presentaban menor dilatación post-IAM que el grupo control; en concreto, el volumen telesistólico (LVESV), el mejor predictor de eventos adversos en remodelado del VI (46,47), era significativamente menor en el grupo tratado con el agonista de S1P-R analizado tanto por RM como por ecocardiografía 3D.
Finalmente, la hipertrofia del cardiomiocito del tejido miocárdico remoto no infartado es un mecanismo compensatorio inicial importante, pero si se prolonga en el tiempo provoca incremento en la masa del VI y se convierte en un proceso negativo asociado a eventos adversos (48). Los efectos cardioprotectores de la activación de S1P-R en el momento del IM redujeron el nivel de remodelado compensatorio del VI, como muestra la menor masa del VI, la menor activación de Akt y ERK1/2 y el menor tamaño del cardiomiocito en los cerdos tratados con Fingolimod un mes post-IAM. Akt y ERK1/2 son importantes inductores del crecimiento y supervivencia de los miocitos; mientras que la activación (fosforilación) aguda y puntual de Akt da lugar a una disminución de la apoptosis, la activación crónica de Akt y de ERK da lugar a hipertrofia, aspecto clave en el remodelado adverso del VI (49) y relacionado con efectos negativos sobre el miocardio (50). Por ello, la disminución de los ratios pAkt/Akt y pErk/Erk en el tejido miocárdico remoto no infartado en los cerdos tratados con Fingolimod, un mes después del IM, es un reflejo del menor remodelado del VI presente en este grupo, mientras que la elevación de pAkt en el miocardio isquémico a las 24 horas post-IAM en el grupo tratado con Fingolimod es el resultado de una mayor activación de las rutas de supervivencia mediadas por S1P-R (resultado consistente con una menos tasa de apoptosis en el grupo Fingolimod). La reducción en la hipertrofia de los cardiomiocitos (tanto tamaño como perímetro) a nivel histológico en el grupo Fingolimod apoya además esta disminución de la hipertrofia del VI. Finalmente, la fibrosis miocárdica intersticial en el miocardio remoto no isquémico, otro marcador de remodelado cardiaco (44,45,51), era menor en los cerdos tratados con Fingolimod, lo que corrobora la prevención de remodelado del VI producida por la activación de S1P-R.
La activación crónica de la respuesta neurohormonal, especialmente del sistema nervioso simpático (52), es un componente principal en el remodelado del VI. De hecho, los niveles plasmáticos de catecolaminas son un importante predictor de la mortalidad cardiovascular en pacientes tras un IAM (53,54). En este estudio, nos hemos enfocado en medir los productos de degradación de las catecolaminas plasmáticas (metanefrinas) ya que presentan una mayor estabilidad y representan la actividad neurohormonal a largo plazo (55). Curiosamente, los niveles plasmáticos de metanefrinas eran menores en el grupo Fingolimod que en el grupo control, lo que indica un menor remodelado post-IAM en los animales tratados con agonismo de S1P-R.
Limitaciones del estudio
Nuestro estudio presenta varias limitaciones. En primer lugar no podemos estar seguros de si los beneficios de Fingolimod en la prevención del remodelado cardiaco post-IAM se deben exclusivamente a la reducción inicial del tamaño de IAM o si la activación de S1P-R per se posee efectos anti-remodelado. De hecho, el tratamiento crónico con Fingolimod previno el remodelado patológico del VI en otro estudio en modelo murino de sobrecarga de presión (56). No obstante, dado que nosotros administramos el fármaco a lo largo de un periodo de tiempo muy limitado (pre-reperfusióny durante 3 días post-IAM), creemos que la prevención del remodelado del VI en nuestro modelo se debe a una reducción del tamaño de IAM y no a un efecto crónico directo de Fingolimod en el miocardio no isquémico. Sin embargo, más estudios son necesarios para responder esta pregunta.
La enfermedad coronaria en humanos está asociada frecuentemente con comorbilidades que no están presentes en este modelo animal como por ejemplo hipertensión, diabetes mellitus o hiperlipidemia. Por otro lado, no sabemos qué potencia podría tener la activación de S1P-R sobre el remodelado cardiaco con las terapias médicas estándar con beta-bloqueantes, inhibidores de la ECA y los antagonistas de la aldosterona. Por lo tanto, la eficacia clínica de la activación de S1P-R en el contexto del IAM tendría que ser investigada en pacientes humanos.
5. CONCLUSIONES
Como conclusión, nuestros datos proporcionan un apoyo al concepto de la intervención farmacológica con el agonista de S1P-R Fingolimod en la reducción de los efectos deletéreos del IAM en un contexto experimental. Hemos demostrado por primera vez que la activación de S1P-R con Fingolimod antes de la reperfusión mejora el miocardio preservado y reduce el tamaño de infarto, dando lugar a una mejora de la función sistólica y de la mecánica del VI y a una reducción del remodelado del VI en un modelo con animales grandes. Estos efectos cardioprotectores de Fingolimod en un contexto agudo están mediados por la activación de la ruta RISK y la reducción de la apoptosis. Estos hallazgos muestran el potencial terapéutico de la señalización de S1P-R en el periodo peri-infarto y justifica futuros estudios en pacientes humanos.
6. referencias
1. De Luca G.; Suryapranata H.; Ottervanger JP.; Antman EM. Time delay to treatment and mortality in primary angioplasty for acute myocardial infarction, every minute of delay counts. Circulation 109(10), 1223-1225 (2004).
2. Yellon DM.; Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 357(11), 1121-1135 (2007).
3. Hausenloy DJ.; Yellon DM. Myocardial ischemia-reperfusion injury, a neglected therapeutic target. J Clin Invest 123(1), 92-100 (2013).
4. Karliner JS. Sphingosine kinase and sphingosine 1-phosphate in the heart, a decade of progress. Biochim Biophys Acta 1831(1), 203-212 (2012).
5. Spiegel S.; Milstien S. Sphingosine-1-phosphate, an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4(5), 397-407 (2003).
6. Man K.; Ng KT.; Lee TK.; Lo CM.; Sun CK.; Li XL.; Zhao Y.; Ho JW.; Fan ST. FTY720 attenuates hepatic ischemia-reperfusion injury in normal and cirrhotic livers. Am J Transplant 5(1), 40-49 (2005).
7. Delbridge MS.; Shrestha BM.; Raftery AT.; El Nahas AM.; Haylor JL. Reduction of ischemia-reperfusion injury in the rat kidney by FTY720.; a synthetic derivative of sphingosine. Transplantation 84(2), 187-195, (2007).
8. Laychock SG.; Sessanna SM.; Lin MH.; Mastrandrea LD. Sphingosine 1-phosphate affects cytokine-induced apoptosis in rat pancreatic islet beta-cells. Endocrinology 147(10), 4705-4712 (2006)
9. Hasegawa Y.; Suzuki H.; Sozen T.; Rolland W.; Zhang JH. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 41(2), 368-374 (2010).
10. Karliner JS.; Honbo N.; Summers K.; Gray MO.; Goetzl EJ. The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J Mol Cell Cardiol 33(9), 1713-1717 (2001).
11. Tao R.; Zhang J.; Vessey DA.; Honbo N.; Karliner JS. Deletion of the sphingosine kinase-1 gene influences cell fate during hypoxia and glucose deprivation in adult mouse cardiomyocytes. Cardiovasc Res 74(1), 56-63 (2007).
12. Jin ZQ.; Zhou HZ.; Zhu P.; Honbo N.; Mochly-Rosen D.; Messing RO.; Goetzl EJ.; Karliner JS.; Gray MO. Cardioprotection mediated by sphingosine-1-phosphate and ganglioside GM-1 in wild-type and PKC epsilon knockout mouse hearts. Am J Physiol Heart Circ Physiol 282(6), H1970-1977 (2002).
13. Lecour S.; Smith RM.; Woodward B.; Opie LH.; Rochette L.; Sack MN. Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J Mol Cell Cardiol 34(5), 509-518 (2002).
14. Duan HF.; Wang H.; Yi J.; Liu HJ.; Zhang QW.; Li LB.; Zhang T.; Lu Y.; Wu CT.; Wang LS. Adenoviral gene transfer of sphingosine kinase 1 protects heart against ischemia/reperfusion-induced injury and attenuates its postischemic failure. Hum Gene Ther 18(11), 1119-1128 (2007).
15. Means CK.; Xiao CY.; Li Z.; Zhang T.; Omens JH.; Ishii I.; Chun J.; Brown JH. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 292(6), H2944-2951 (2007)
16. Brinkmann V.; Billich A.; Baumruker T.; Heining P.; Schmouder R.; Francis G.; Aradhye S.; Burtin P. Fingolimod (FTY720), discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 9(11), 883-897 (2010)
17. Kappos L.; Radue EW.; O'Connor P.; Polman C.; Hohlfeld R.; Calabresi P.; Selmaj K.; Agoropoulou C.; Leyk M.; Zhang-Auberson L.; Burtin P. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 362(5), 387-401 (2010).
18. Cohen JA.; Barkhof F.; Comi G.; Hartung HP.; Khatri BO.; Montalban X.; Pelletier J.; Capra R.; Gallo P.; Izquierdo G.; Tiel-Wilck K.; de Vera A.; Jin J.; Stites T.; Wu S.; Aradhye S.; Kappos L. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 362(5), 402-415 (2010)
19. Zhang J.; Honbo N.; Goetzl EJ.; Chatterjee K.; Karliner JS.; Gray MO. Signals from type 1 sphingosine 1-phosphate receptors enhance adult mouse cardiac myocyte survival during hypoxia. Am J Physiol Heart Circ Physiol 293(5), H3150-3158 (2007).
20. Hofmann U.; Burkard N.; Vogt C.; Thoma A.; Frantz S.; Ertl G.; Ritter O.; Bonz A. Protective effects of sphingosine-1-phosphate receptor agonist treatment after myocardial ischaemia-reperfusion. Cardiovasc Res 83(2), 285-293 (2009).
21. Egom EE.; Mohamed TM.; Mamas MA.; Shi Y.; Liu W.; Chirico D.; Stringer SE.; Ke Y.; Shaheen M.; Wang T.; Chacko S.; Wang X.; Solaro RJ.; Fath-Ordoubadi F.; Cartwright EJ.; Lei M. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am J Physiol Heart Circ Physiol 301(4), H1487-1495 (2011).
22. Ibanez B.; Prat-Gonzalez S.; Speidl WS.; Vilahur G.; Pinero A.; Cimmino G.; Garcia MJ.; Fuster V.; Sanz J.; Badimon JJ. Early metoprolol administration before coronary reperfusion results in increased myocardial salvage, analysis of ischemic myocardium at risk using cardiac magnetic resonance. Circulation 115(23), 2909-2916 (2007).
23. Aletras AH.; Tilak GS.; Natanzon A.; Hsu LY.; Gonzalez FM.; Hoyt RF.; Jr..; Arai AE. Retrospective determination of the area at risk for reperfused acute myocardial infarction with T2-weighted cardiac magnetic resonance imaging, histopathological and displacement encoding with stimulated echoes (DENSE) functional validations. Circulation 113(15), 1865-1870 (2006).
24. Lang RM.; Bierig M.; Devereux RB.; Flachskampf FA.; Foster E.; Pellikka PA.; Picard MH.; Roman MJ.; Seward J.; Shanewise JS.; Solomon SD.; Spencer KT.; Sutton MS.; Stewart WJ. Recommendations for chamber quantification, a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group.; developed in conjunction with the European Association of Echocardiography.; a branch of the European Society of Cardiology. J Am Soc Echocardiogr 18(12), 1440-1463 (2005)
25. Mor-Avi V.; Lang RM.; Badano LP.; Belohlavek M.; Cardim NM.; Derumeaux G.; Galderisi M.; Marwick T.; Nagueh SF.; Sengupta PP.; Sicari R.; Smiseth OA.; Smulevitz B.; Takeuchi M.; Thomas JD.; Vannan M.; Voigt JU.; Zamorano JL. Current and evolving echocardiographic techniques for the quantitative evaluation of cardiac mechanics, ASE/EAE consensus statement on methodology and indications endorsed by the Japanese Society of Echocardiography. J Am Soc Echocardiogr 24(3), 277-313 (2011).
26. Savu O.; Jurcut R.; Giusca S.; van Mieghem T.; Gussi I.; Popescu BA.; Ginghina C.; Rademakers F.; Deprest J.; Voigt JU. Morphological and functional adaptation of the maternal heart during pregnancy. Circ Cardiovasc Imaging 5(3), 289-297 (2012).
27. Cuvillier O.; Pirianov G.; Kleuser B.; Vanek PG.; Coso OA.; Gutkind S.; Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381(6585), 800-803 (1996).
28. Jin ZQ.; Zhang J.; Huang Y.; Hoover HE.; Vessey DA.; Karliner JS. A sphingosine kinase 1 mutation sensitizes the myocardium to ischemia/reperfusion injury. Cardiovasc Res 76(1), 41-50 (2007).
29. Jin ZQ.; Goetzl EJ.; Karliner JS. Sphingosine kinase activation mediates ischemic preconditioning in murine heart. Circulation 110(14), 1980-1989 (2004).
30. Theilmeier G.; Schmidt C.; Herrmann J.; Keul P.; Schafers M.; Herrgott I.; Mersmann J.; Larmann J.; Hermann S.; Stypmann J.; Schober O.; Hildebrand R.; Schulz R.; Heusch G.; Haude M.; von Wnuck Lipinski K.; Herzog C.; Schmitz M.; Erbel R.; Chun J.; Levkau B. High-density lipoproteins and their constituent.; sphingosine-1-phosphate.; directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 114(13), 1403-1409 (2006).
31. Piot C.; Croisille P.; Staat P.; Thibault H.; Rioufol G.; Mewton N.; Elbelghiti R.; Cung TT.; Bonnefoy E.; Angoulvant D.; Macia C.; Raczka F.; Sportouch C.; Gahide G.; Finet G.; Andre-Fouet X.; Revel D.; Kirkorian G.; Monassier JP.; Derumeaux G.; Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359(5), 473-481 (2008).
32. Ovize M.; Baxter GF.; Di Lisa F.; Ferdinandy P.; Garcia-Dorado D.; Hausenloy DJ.; Heusch G.; Vinten-Johansen J.; Yellon DM.; Schulz R. Postconditioning and protection from reperfusion injury, where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res 87(3), 406-423 (2010).
33. Kwong RY.; Chan AK.; Brown KA.; Chan CW.; Reynolds HG.; Tsang S.; Davis RB. Impact of unrecognized myocardial scar detected by cardiac magnetic resonance imaging on event-free survival in patients presenting with signs or symptoms of coronary artery disease. Circulation 113(23), 2733-2743 (2006).
34. Cheong BY.; Muthupillai R.; Wilson JM.; Sung A.; Huber S.; Amin S.; Elayda MA.; Lee VV.; Flamm SD. Prognostic significance of delayed-enhancement magnetic resonance imaging, survival of 857 patients with and without left ventricular dysfunction. Circulation 120(21), 2069-2076 (2009).
35. Yan AT.; Shayne AJ.; Brown KA.; Gupta SN.; Chan CW.; Luu TM.; Di Carli MF.; Reynolds HG.; Stevenson WG.; Kwong RY. Characterization of the peri-infarct zone by contrast-enhanced cardiac magnetic resonance imaging is a powerful predictor of post-myocardial infarction mortality. Circulation 114(1), 32-39 (2006).
36. Kelle S.; Roes SD.; Klein C.; Kokocinski T.; de Roos A.; Fleck E.; Bax JJ.; Nagel E. Prognostic value of myocardial infarct size and contractile reserve using magnetic resonance imaging. J Am Coll Cardiol 54(19), 1770-1777 (2009).
37. Wu E.; Ortiz JT.; Tejedor P.; Lee DC.; Bucciarelli-Ducci C.; Kansal P.; Carr JC.; Holly TA.; Lloyd-Jones D.; Klocke FJ.; Bonow RO. Infarct size by contrast enhanced cardiac magnetic resonance is a stronger predictor of outcomes than left ventricular ejection fraction or end-systolic volume index, prospective cohort study. Heart 94(6), 730-736 (2008).
38. Masci PG.; Ganame J.; Strata E.; Desmet W.; Aquaro GD.; Dymarkowski S.; Valenti V.; Janssens S.; Lombardi M.; Van de Werf F.; L'Abbate A.; Bogaert J. Myocardial salvage by CMR correlates with LV remodeling and early ST-segment resolution in acute myocardial infarction. JACC Cardiovasc Imaging 3(1), 45-51 (2010).
39. Solomon SD.; Anavekar N.; Skali H.; McMurray JJ.; Swedberg K.; Yusuf S.; Granger CB.; Michelson EL.; Wang D.; Pocock S.; Pfeffer MA. Influence of ejection fraction on cardiovascular outcomes in a broad spectrum of heart failure patients. Circulation 112(24), 3738-3744 (2005).
40. Williams MJ.; Odabashian J.; Lauer MS.; Thomas JD.; Marwick TH. Prognostic value of dobutamine echocardiography in patients with left ventricular dysfunction. J Am Coll Cardiol. 27(1), 132-139 (1996).
41. Geyer H.; Caracciolo G.; Abe H.; Wilansky S.; Carerj S.; Gentile F.; Nesser HJ.; Khandheria B.; Narula J.; Sengupta PP. Assessment of myocardial mechanics using speckle tracking echocardiography, fundamentals and clinical applications. Journal of the American Society of Echocardiography , official publication of the American Society of Echocardiography 23(4), 351-369, quiz 453-355 (2010).
42. Stanton T.; Leano R.; Marwick TH. Prediction of all-cause mortality from global longitudinal speckle strain, comparison with ejection fraction and wall motion scoring. Circulation. Cardiovascular imaging 2(5), 356-364 (2009).
43. Hung CL.; Verma A.; Uno H.; Shin SH.; Bourgoun M.; Hassanein AH.; McMurray JJ.; Velazquez EJ.; Kober L.; Pfeffer MA.; Solomon SD.; investigators V. Longitudinal and circumferential strain rate.; left ventricular remodeling.; and prognosis after myocardial infarction. Journal of the American College of Cardiology 56(22), 1812-1822 (2010).
44. Konstam MA.; Kramer DG.; Patel AR.; Maron MS.; Udelson JE. Left ventricular remodeling in heart failure, current concepts in clinical significance and assessment. JACC Cardiovasc Imaging 4(1), 98-108 (2011).
45. Cohn JN.; Ferrari R.; Sharpe N. Cardiac remodeling--concepts and clinical implications, a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. Journal of the American College of Cardiology 35(3), 569-582 (2000).
46. White HD.; Norris RM.; Brown MA.; Brandt PW.; Whitlock RM.; Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation 76(1), 44-51 (1987).
47. Migrino RQ.; Young JB.; Ellis SG.; White HD.; Lundergan CF.; Miller DP.; Granger CB.; Ross AM.; Califf RM.; Topol EJ. End-systolic volume index is a strong predictor of early and late mortality. The GUSTO-I Angiographic Investigators. Circulation 96(1), 116-121 (1997).
48. Verma A.; Meris A.; Skali H.; Ghali JK.; Arnold JM.; Bourgoun M.; Velazquez EJ.; McMurray JJ.; Kober L.; Pfeffer MA.; Califf RM.; Solomon SD. Prognostic implications of left ventricular mass and geometry following myocardial infarction, the VALIANT (VALsartan In Acute myocardial iNfarcTion) Echocardiographic Study. JACC Cardiovasc Imaging 1(5), 582-591 (2008).
49. van Berlo JH.; Maillet M.; Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest 123(1), 37-45 (2013).
50. Haq S.; Choukroun G.; Lim H.; Tymitz KM.; del Monte F.; Gwathmey J.; Grazette L.; Michael A.; Hajjar R.; Force T.; Molkentin JD. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 103(5), 670-677 (2001).
51. Gonzalez A.; Ravassa S.; Beaumont J.; Lopez B.; Diez J. New targets to treat the structural remodeling of the myocardium. J Am Coll Cardiol 58(18), 1833-1843 (2011).
52. Lymperopoulos A.; Rengo G.; Koch WJ. Adrenergic nervous system in heart failure, pathophysiology and therapy. Circ Res 113(6), 739-753 (2013).
53. Francis GS.; Cohn JN.; Johnson G.; Rector TS.; Goldman S.; Simon A. Plasma norepinephrine.; plasma renin activity.; and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. The V-HeFT VA Cooperative Studies Group. Circulation. 87(6 Suppl), VI40-48 (1993).
54. Benedict CR.; Shelton B.; Johnstone DE.; Francis G.; Greenberg B.; Konstam M.; Probstfield JL.; Yusuf S. Prognostic significance of plasma norepinephrine in patients with asymptomatic left ventricular dysfunction. SOLVD Investigators. Circulation. 94(4), 690-697 (1996).
55. Raake PW.; Schlegel P.; Ksienzyk J.; Reinkober J.; Barthelmes J.; Schinkel S.; Pleger S.; Mier W.; Haberkorn U.; Koch WJ.; Katus HA.; Most P.; Muller OJ. AAV6.betaARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur Heart J 34(19), 1437-1447 (2013).