ARTÍCULO |
Protein tyrosine phosphatase deficiency protects against the induction of the early apoptosis by paracetamol in hepatocytes
Maysa Ahmed Mobasher*, Ángela María Valverde
Instituto de Investigaciones Biomédicas Alberto Sols (CSIC-UAM), 28029 Madrid, Spain.
*e-mail: maysaonline@yahoo.com
An. Real Acad. Farm. Vol. 80, Nº 4 (2014), pag.666-682
Premio “Consejo General de Colegios Oficiales de Farmacéuticos” en el Concurso Científico 2013 de la Real Academia Nacional de Farmacia.
abstract
Acetaminophen (paracetamol, APAP) is a widely used analgesic and antipyretic drug safe at therapeutic doses but its overdose causes liver injury. Our goal was to explore whether protein tyrosine phosphatase 1B (PTP1B), a negative modulator of survival signaling pathways, plays a role in APAP-induced cell death in hepatocytes. Hepatotoxicity was evaluated in immortalized hepatocytes generated from wild-type (PTP1B+/+) and PTP1B-deficient (PTP1B-/-) mice. Apoptosis occurred as an early event only in APAP-treated PTP1B+/+ hepatocytes. PTP1B deficiency conferred protection against cell cycle arrest and loss of cellular viability. These data suggest that PTP1B as a target against APAP-induced liver failure. |
Keywords: Paracetamol; PTP1B; apoptosis.
resumen
La inhibición de la proteína tirosina fosfatasa 1B protege frente a la apoptosis temprana inducida por paracetamol en hepatocitos
El paracetamol es un analgésico/antipirético hepatotóxico a dosis altas. Investigamos el papel de la proteína tirosina fosfatasa 1B (PTP1B), un modulador negativo de señalización de supervivencia celular, en la muerte celular temprana (apoptosis) inducida por paracetamol en hepatocitos. En hepatocitos controles se inducía apoptosis en respuesta al paracetamol. Este efecto se encontraba disminuido en hepatocitos deficientes en PTP1B. La falta de PTP1B protegía a los hepatocitos de la parada del ciclo celular y la pérdida de la viabilidad celular tras el tratamiento con paracetamol. Proponemos a la PTP1B como diana terapéutica frente al fallo hepático inducido por sobredosis de paracetamol. |
Palabras clave: Paracetamol, PTP1B, apoptosis.
1. INTRODUCTION
Acetaminophen (APAP) or Paracetamol is a widely used analgesic and antipyretic drug safe at therapeutic doses (1). However, an accidental or intentional overdose can induce severe hepatotoxicity in both experimental animals and humans (2). The first reports of APAP hepatotoxicity in humans appeared in the literature in the 1960s. In fact, APAP overdose is the most frequent cause of drug-induced liver failure in the United States and in Great Britain at present (3). Therefore, APAP-induced acute toxicity has become an essential model for studying drug-induced liver and kidney failure. In the liver, APAP overdose produces a centrilobular hepatic necrosis that can be fatal and is increasingly recognized as a significant public health problem (4-6). Moreover, APAP overdose is also the second leading cause of liver transplantation, which accounts for considerable levels of morbidity and mortality (7).
APAP is commonly used for the relief of minor pains like headaches and in combination with opioid analgesics is also used in the management of more severe pains in advanced cancer and in post-operative periods. While APAP has analgesic and antipyretic properties comparable to those of aspirin, its anti-inflamatory effects are weak. The mechanisms underlying APAP-induced liver injury have been studied for several decades and excellent recent reviews have revealed the main cellular and molecular pathways involved in its toxic response (8-12). However, despite of substantial progress in our understanding of APAP-induced hepatotoxicity, additional mechanisms responsible of the cellular damage induced by this drug remain still unknown.
The initial step in APAP-mediated toxicity is initiated by cytochrome P-450 (CYP) by a direct two electron oxidation of APAP, a previously unrecognized mechanism of CYP P450-mediated reactions, that convert APAP to the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) causing glutathione (GSH) depletion and covalent binding to hepatic parenchymal cell proteins and DNA (13). The CYP isoforms important in APAP metabolism are CYP2E1, CYP1A2, CYP3A4 and CYP2D6 (14-15). Moreover, generation of reactive oxygen (ROS) and nitrogen species, lipid peroxidation, mitochondrial dysfunction, disruption of calcium homeostasis and induction of apoptosis and necrosis are also involved in APAP-induced hepatotoxicity (16).
At therapeutic doses, NAPQI is efficiently detoxified by GSH and eliminated in the urine or bile as APAP–cysteine, APAP–N-acetylcysteine (APAP–NAC), and APAP–glutathione (APAP–GSH). After an overdose of APAP, the glucuronidation and sulfation routes become saturated and more extensive bioactivation of the drug occurs leading to rapid depletion of the hepatic GSH pool. Subsequently, NAPQI binds to cysteine groups on cellular proteins forming APAP-protein adducts (17). Of note, NAPQI also binds to a number of mitochondrial proteins (9-10) which, in turn, causes oxidative stress that may trigger signaling pathways through mitochondrial toxicity ultimately leading to lethal cell injury.
As stated above, the precise mechanisms by which APAP mediates hepatotoxicity in both humans and experimental animals still remain to be elucidated. Metabolic activation of APAP and NAPQI binding to target proteins and DNA seem to be necessary but are not sufficient for toxicity. Recent findings indicate that a combination of mitochondrial oxidative stress, increased calcium levels and other factors may trigger the mitochondrial membrane permeability transition (MPT) pore opening resulting in the collapse of the transmembrane potential and then osmotic swelling. These events ultimately cause the rupture of the outer mitochondrial membrane and release of cytochrome c and other pro-apoptotic factors into the cytosol (18).
Regarding the signaling pathways mediated by various growth factors and hormones committed in the regulation of cellular metabolism, differentiation, growth and survival, protein tyrosine phosphorylation constitutes a key element. Given the importance of tyrosine kinase signaling in maintaining birth and death rates of cells, these pathways must be regulated carefully. Protein tyrosine phosphatases (PTPs) catalyze the dephosphorylation of tyrosine-phosphorylated proteins (19) and are known to be important negative regulators of growth factor signaling. Several PTPs have been described which may participate in modulating the balance between survival and cell death. Among them, protein tyrosine phosphatase 1B (PTP1B) is a widely expressed non-receptor PTP that is associated with the endoplasmic reticulum (ER) and other intracellular membranes via a hydrophobic interaction of its C-terminal targeting sequence (20). In particular, PTP1B dephosphorylates and inactivates receptors belonging to the tyrosine kinase superfamily such as the EGF receptor (EGFR) (21), the PDGFR (22), the insulin receptor (IR) (23) and the IGF-IR (24), all of them implicated in the survival of hepatocytes (25-26) and many other cellular models. Thus, PTPs represent novel molecular targets for the development of medicinal reagents that possess distinct modes of action.
During the last years, our laboratory has been interested in the study of key proteins that mediate survival pathways in hepatocytes. In this regard, the precise role of PTP1B in these processes has been investigated in a study published by Gonzalez-Rodriguez et al. (27) which was performed in immortalized neonatal hepatocyte cell lines from wild-type (PTP1B+/+) and PTP1B-deficient (PTP1B-/-) mice. These cell lines have been proven to be excellent tools for in vitro studies of cell death/survival because they express endogenous pro- and anti-apoptotic proteins at levels comparable to the liver and hepatocytes of neonatal mice. Using these cell lines it was demonstrated that the lack of PTP1B protects against apoptosis induced by trophic factors withdrawal, whereas increased expression of this phosphatase sensitizes neonatal hepatocytes to death signals induced by this apoptotic stimulus (27). These differential effects resulted from the regulation of multiple events involving mitochondrial integrity, the Bcl-2 protein family, the caspases-8,-9 and -3 and the nuclear translocation of Foxo 1 which activates the death receptor pathway. Thus, these previous observations suggested that levels of PTB1B may exert a pivotal role in maintaining the balance between survival and death in hepatocytes.
On that basis, the main goal of the present study is to investigate if PTP1B deficiency is able to protect hepatocytes against the early events during APAP-induced hepatotoxicity.
2. materials and methods
2.1. Materials.
Fetal serum (FS) and culture reagents were obtained from Invitrogen. APAP was purchased from Sigma (Sigma-Aldrich). Anti-phospho-JNK (#9251) (Thr183/ Tyr185) and anti-active caspase 3 (#9661) antibodies were from Cell Signaling Technology. The antibodies against phospho-Akt (Ser 473) (sc-7985), phospho-IGF-IR (Tyr 1165/1166) (sc-101704) and total JNK (sc-571) were from Santa Cruz Biotechnology. The anti-IRS-1 (06-248), anti-IRS-2 (06-248) and anti-mouse PTP1B (07-088) antibodies were obtained from Upstate (Millipore). Anti-BclxL (610211) antibody was from BD Pharmingen. Anti-β-actin (A-5441) antibody was from Sigma. Anti-Cyp2E1 antibody (aB19140) was from Abcam. Total Akt and total IGF-IR antibodies were gifts from M Birnbaum and MF White, respectively (Joslin Diabetes Center, USA). Anti GCLc and GCLm antibodies were a gift from T Kavanagh (University of Washington, USA).
2.2. Human liver biopsies.
Human liver samples were obtained from from five patients intoxicated with APAP and three healthy subjects. These samples were kindly donated from Dr Kenneth J. Simpson in the division of Clinical and Surgical Sciences, University of Edinburgh, Edinburgh EH164TJ, UK. Informed written consent was obtained from each patient.
2.3. Animal models.
Three months-old male PTP1B+/+ (wild-type) and PTP1B-/- mice maintained on the C57Bl/6J x 129Sv/J genetic background (28) were used throughout the study. Animal experimentation was conducted accordingly to the accepted guidelines for animal care of the Comunidad de Madrid (Spain). Overnight fasted mice were intraperitoneally (i.p.) injected with 500 mg/kg APAP dissolved in physiological saline. Mice were sacrificed at 6 h and livers were collected.
2.4. Liver histology.
Histological grading of hepatic necrosis was performed by two blinded oervbsers using hematoxilin and eosin (H&E)-stained sections as follows: 30% of the total area necrotic (1 point); 30–60% of the total area necrotic (2 points); 60% of the total area necrotic (3 points).
2.5. Generation of immortalized hepatocyte cell lines.
The detailed protocol for the generation of immortalized neonatal hepatocytes from wild-type and PTP1B-/- mice is described in (27). For re-expression of PTP1B in deficient cells, PTP1B-/- neonatal hepatocytes were reconstituted with retroviral Myc-tagged PTP1B (kindly provided by M. L. Tremblay, McGill Cancer Center, Quebec, Canada) and four pools of infected cells were selected with hygromycin B (200 μg/ml) for 2 weeks. As a control, PTP1B-/- were infected with an empty vector (pBabe hygro). The expression of PTP1B in the different cell lines was assessed by Western blot.
2.6. Isolation and culture of primary mouse hepatocytes.
Mouse hepatocytes were isolated from male mice (8-12 weeks-old) by perfusion with collagenase and cultured as described (27).
2.7. PTP1B immunohistochemistry.
PTP1B expression was analyzed by immunohistochemistry in human liver samples as previously described (29).
2.8. Determination of Reactive Oxygen Species (ROS).
Cellular ROS were quantified by flow cytometry using the dichlorofluorescin (DCFH) probe. APAP-stimulated cells were detached by trisinization and collected by centrifugation at 2.500 x g for 4 min. Then, cells were washed and resuspended in 500 microliters of PBS. DCFH (10 μM) and propidium iodide (0,002% w/v) were added 10 min before measurement. Fluorescence was measured with 488-nm laser excitation and 510-nm for emission.
2.9. Analysis of caspase-3 activity.
At the end of the treatments, cells were scraped off, collected by centrifugation at 2.500 x g for 5 min and lysed at 4ºC in 5 mM Tris/HCl pH 8, 20 mM EDTA, 0,5% Triton X-100. Lysates were clarified by centrifugation at 13.000 x g for 10 min. Reaction mixtures contained 25 microliters of cell lysate, 325 microliters of assay buffer (20 mM HEPES pH 7.5, 10% glycerol, 2 mM dithiothreitol) and 20 µM caspase-3 substrate (Ac-DEVD-AMC). After 2-hour incubation at 37ºC in the dark, enzymatic activity was measured in a luminescence spectrophotometer (Perkin Elmer LS-50, Norwalk, CT) ( excitation, 380 nm; emission, 440 nm). We define a unit of caspase-3 activity as the amount of active enzyme necessary to produce an increase in 1 arbitrary unit in the fluorimeter after 2-hour incubation with the reaction mixture. Protein concentration of cell lysates was determined and the results are presented as caspase-3 activity/micrograms of total protein.
2.10. Cell viability and cytotoxicity.
Cell viability/damage was determined by two alternative methods: gross detection of cell viability by using the crystal violet assay (30) and cytotoxicity assay by measuring lactate dehydrogenase (LDH) leakage into the extracellular medium (31). For the crystal violet assay, cells were seeded at low density (104 cells per well) in 24-well plates, grown for 12 hours with the different treatments and incubated with crystal violet (0.2% in ethanol) for 20 min. Plates were rinsed with distilled water and allowed to dry and 1% SDS was then added. The absorbance of each well was measured using a microplate reader at 570 nm (Bio-Tek, Winooski, VT, USA). Cytotoxicity was evaluated by the LDH method collecting both the culture medium and cells that were scraped in phosphate-buffered saline (PBS) after the different treatments. Then, cells were sonicated to ensure breaking down the cell membrane to release the total amount of LDH followed by centrifugation (1.000 ×g 15 min) to clear up the cell sample. 11 microliters of extract were placed into a well of a 96-multiwell system for the assay. In the same manner, 11 microliters of each culture medium sample were also deposited in each 96-multiwell. The LDH leakage was estimated from the ratio between the LDH activity in the culture medium and that of the whole cell content. Fluorescence was measured at an emission wavelength of 460 nm and an excitation wavelength of 340 nm.
2.11. Quantification of apoptotic cells by flow cytometry.
After APAP stimulation, adherent and non-adherent cells were collected by centrifugation, washed with PBS and fixed with cold ethanol. The cells were then washed, resuspended in PBS, and incubated with RNAse A (25 µg/106 cells) for 30 min at 37ºC. After addition of 0.05% propidium iodide, cells were analyzed by flow cytometry.
2.12. Analysis of alanine amino transaminase (ALT) activity.
Blood was collected in tubes containing heparin and diluted 1/30 with saline (0.9% NaCl). ALT activity was determined by direct measurement with the Reflotron test (Ref. 10745120, Roche Diagnostics).
2.13. Protein determination.
Protein determination was performed by the Bradford dye method, using the Bio-Rad reagent and BSA as the standard.
2.14. Western blot analysis.
To obtain total cell lysates, cells from supernatants were collected by centrifugation at 2.000 x g for 5 min at 4ºC. Attached cells were scraped off in ice-cold PBS, pelleted by centrifugation at 4.000 x g for 10 min at 4ºC and resuspended in lysis buffer (25 mM HEPES, 2.5 nM EDTA, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride and 5 µg/ml leupeptin). Cellular lysates were clarified by centrifugation at 12.000 x g for 10 min. After SDS-PAGE, gels were transferred to Immobilon membranes (Millipore), blocked using 5% non-fat dried milk or 3% bovine serum albumin (BSA) in 10 mM Tris-HCl, 150 mM NaCl pH 7.5 (TBS) and incubated overnight with antibodies as indicated in 0.05% Tween-20-TBS. Immunoreactive bands were visualized using the ECL Western blotting protocol (Millipore).
2.15. Statistical Analysis.
The data are expressed as means ± SD. The statistical significance was estimated with Student's test for unpaired observation (p*<0.05; p**<0.01; and p***<0.001).
3. results and discussion
PTP1B expression is induced in human liver during APAP intoxication.
Cell proliferation and cell death are governed by stimulatory and inhibitory signals. Whereas trophic factors simultaneously stimulate mitosis and inhibit cell death, negative growth signals regulate the opposite of these biological effects. In the liver, trophic factors include endogenous growth factors such as EGF, bFGF, TGF-alpha and IGFs that act through receptors belonging to the tyrosine kinase superfamily (32). Thus, tyrosine phosphorylation may play a regulatory role in the induction and execution of programmed cell death in the liver. On that basis, our first goal in this work was to investigate whether an overdose of APAP induced the expression of PTP1B, a negative modulator of survival-mediated signaling pathways, in human liver biopsies obtained from individuals suffering from APAP intoxication. In our previous work (33) we showed elevated PTP1B expression in patients with APAP toxcicity that needed liver tranplantion. In agreement with this, Figure 1 shows elevated PTP1B expression in another patient suffering from APAP intoxication. Notably, immunostaining was detected in surviving hepatocytes in the areas surrounding the central veins (right panel) as compared to a normal liver (left panel).
PTP1B deficiency protects mouse hepatocytes against elevation of ROS.
The fact that PTP1B has been related to the induction of apoptosis in hepatocytes under conditions of serum withdrawal (27) or activation of the Fas death receptor (34) prompted us to investigate the involvement of PTP1B in the susceptibility of hepatocytes to undergo apoptotic cell death induced by APAP. As indicated in the introduction section, APAP overdoses cause severe hepatotoxicity leading to liver failure in experimental animals and humans. APAP hepatotoxicity is, in part, the result of a series of events that increase cellular oxidative stress. In the liver, Cyp2e1 converts APAP to NAPQI that rapidly depletes GSH and the subsequent generation of ROS (7), and therefore the degree of GSH consumption is a biomarker for APAP bioactivation (35). Both the catalytic and the modifier subunits of γ-glutamyl cysteine ligase (GCL-C and GCL-M) are responsible for glutathione synthesis. Since the expression of Cyp2e1, GCL-C and GCL-M did not change in primary and immortalized hepatocytes from both genotypes of mice (Figure 2A), we used immortalized cells for further experiments.
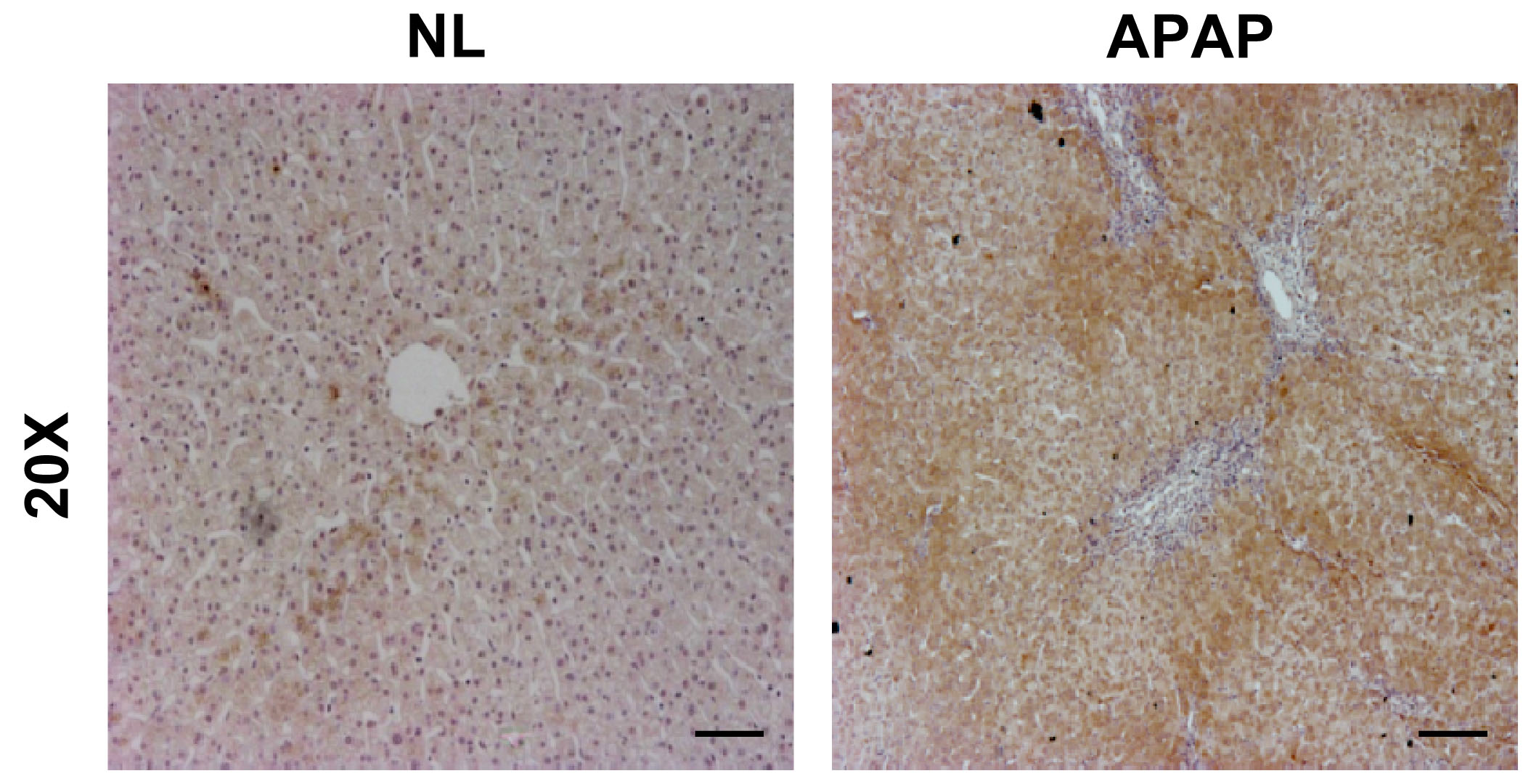
Figure 1.-PTP1B expression is increased during APAP-induced liver injury. Representative anti-PTP1B immunostaining of liver biopsy sections from an individuals with histologically normal liver (NL) or with APAP overdose intoxication (APAP). Bar scale 50 µm.
It has been extensively reported that APAP hepatotoxicity concurs with elevated ROS (36). To evaluate the degree of cellular oxidative stress in APAP-treated hepatocytes, the intracellular ROS production was estimated. Figure 2B shows a representative plot with the shift of the mean fluorescence after APAP treatment for 6 h at 0.5 and 1 mM concentrations in immortalized hepatocytes. Importantly, ROS production was significantly ameliorated in hepatocytes lacking PTP1B compared to the wild-type controls.
Programmed cell death (apoptosis) induced by APAP treatment involves activation of caspase-3: protective effect of PTP1B deficiency.
To further investigate whether caspase-3 is involved in APAP-induced apoptosis, we examined caspase-3 activity in wild-type and PTP1B-/- immortalized hepatocytes. Cells were treated with APAP in a dose-dependent manner and caspase-3 enzymatic activity was analyzed as described in Materials and Methods. As shown in Figure 3A, caspase-3 activity increased with APAP treatment for 8 h in wild-type hepatocytes with a maximal effect at 1 mM concentration. However, in the absence of PTP1B, hepatocytes were protected against activation of caspase-3 upon APAP treatment.
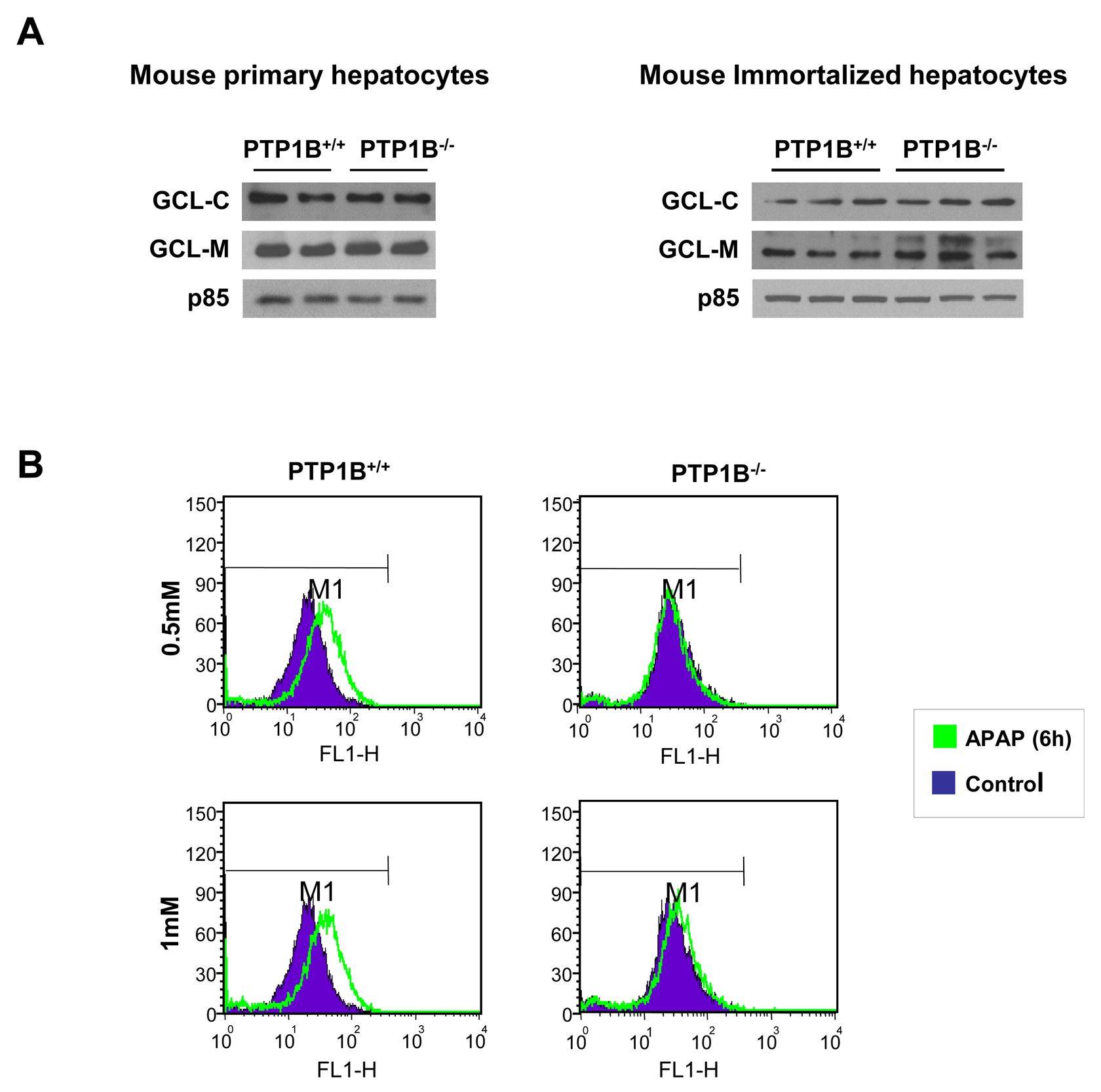
Figure 2.- Effects of PTP1B deficiency in APAP-mediated effects in oxidative stress in hepatocytes. A. Whole cell lysates from PTP1B+/+ and PTP1B-/- mouse primary (left panel) and immortalized (right panel) hepatocytes were analyzed by Western blot with the indicated antibodies. Representative autoradiograms are shown. B. Analysis of ROS levels in APAP-treated wild-type and PTP1B-/- immortalized hepatocytes for 6 h. Representative plots with the shift of the mean fluorescence after APAP treatment are shown.
Caspases are synthesized as inactive zymogens, whose cleavage represents its activation (37). Accordingly, we examined the presence of the active fragment of caspase-3 (15-17 kDa) by Western blot analysis. As shown in Figure 3B, a marked increase in active caspase-3 fragment was observed in APAP-treated wild-type immortalized hepatocytes for 8 h in a dose-dependent manner. However, in PTP1B-/- cells this effect was ameliorated. Importantly, at this early time point we could not see any decrease in the cellular viability as assessed by crystal violet staining and the analysis of LDH enzymatic activity suggesting that apoptosis is a very early event triggered by APAP in hepatocytes (Figure 4A, 4B).
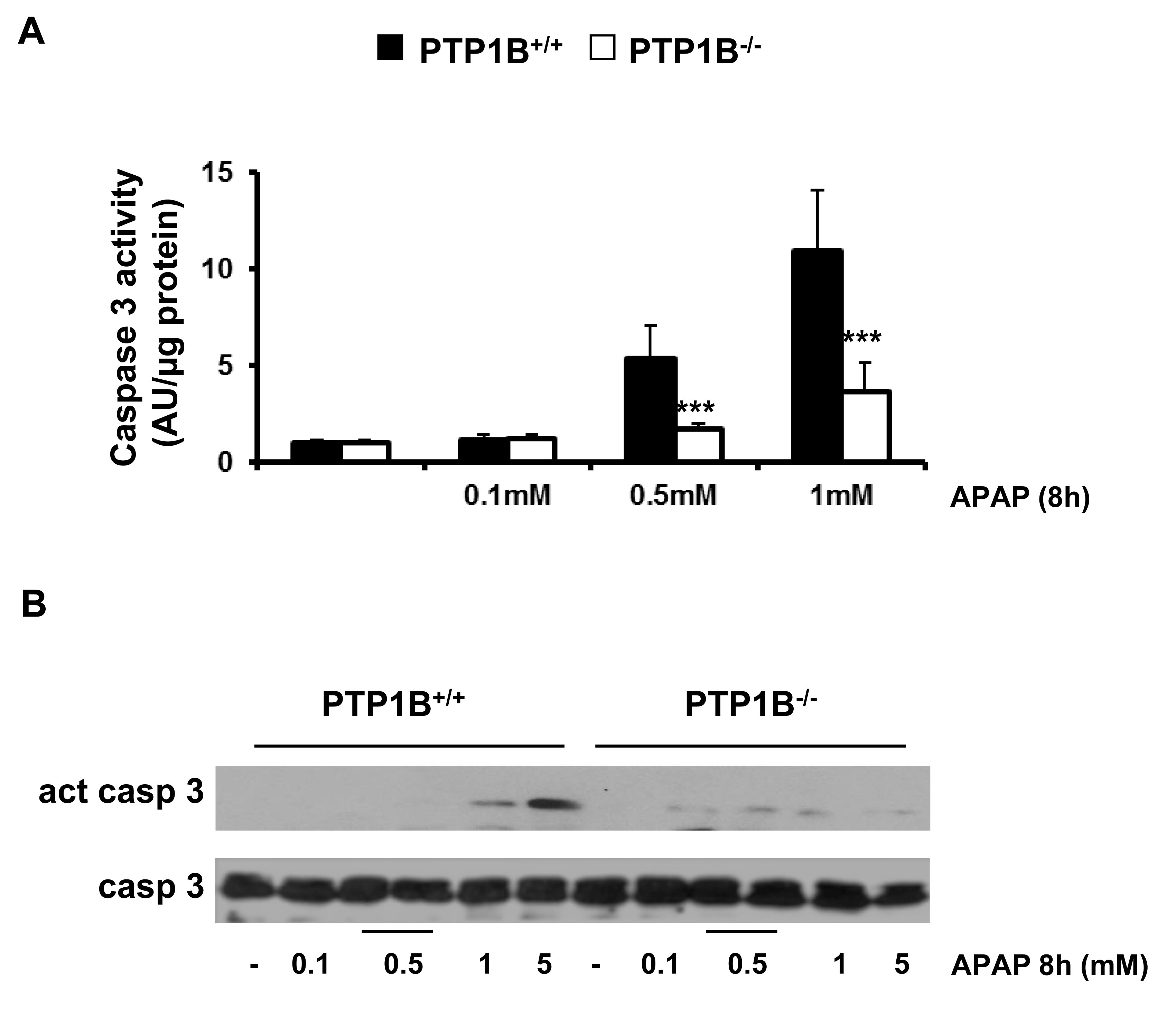
Figure 3.- PTP1B-deficient hepatocytes are protected against APAP-induced caspase-3 activation. PTP1B+/+ and PTP1B-/- immortalized hepatocytes were treated with various doses of APAP for 8 h. A. Analysis of caspase-3 enzymatic activity. B. Analysis of the active caspase-3 fragment in total cell lysates by Western blot. *P<0.05 and ***P<0.005 PTP1B-/- vs. PTP1B+/+ cells (n=4 independent experiments).
Effect of APAP treatment in the cell cycle of wild-type and PTP1B-/- hepatocytes.
We next investigated the effects of APAP in the distribution of the hepatocytes along the phases of the cell cycle including the hypodiploid (sub G0/G1) population. In wild-type hepatocytes, APAP treament induced cell cycle arrest in S phase with a maximal effect at 0.5 mM dose. Of note, under these experimental conditions a twofold increase in the percentage of S phase arrested cells was observed (Figure 5A). This effect was significantly ameliorated in hepatocytes lacking PTP1B. We also observed that APAP treatment increased the percentage of hypodiploid cells in wild-type hepatocytes, but again this effect was significantly reduced in PTP1B-/- hepatocytes (Figure 5B). This result indicates an apoptotic effect of APAP which was significantly reduced in hepatocytes lacking PTP1B.
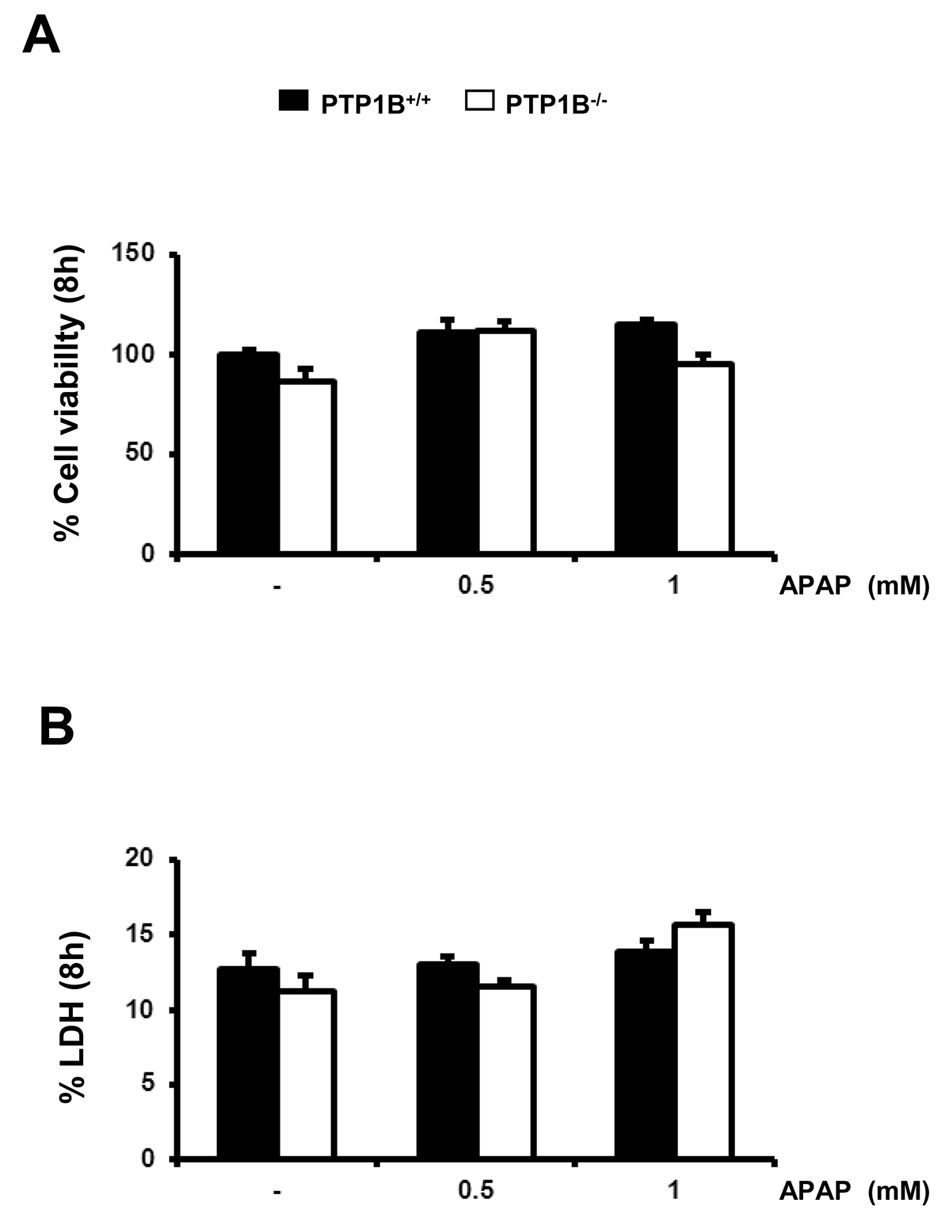
Figure 4.- Immortalized PTP1B-deficient hepatocytes are protected against APAP-induced cell death. PTP1B+/+ and PTP1B-/- immortalized hepatocytes were treated with various doses of APAP for 8 h. Cellular viability (A) and released LDH activity (B) were analyzed. *P<0.05, **P<0.01 and ***P<0.005 PTP1B-/- vs. PTP1B+/+ hepatocytes (n=4 independent experiments).
Cell death (irreversible loss of vital cellular structure and function) is a fundamental phenomenon of biological organisms. Several lines of investigation have led to the concept that there are two fundamental types of cell death: apoptosis and necrosis. The findings of our study demonstrated that the administration of APAP at heptotoxic doses led to the induction of cell death by both necrosis and apoptosis, with apoptotic cell death typically preceeding necrosis. Therefore, we analyzed the population of apoptotic and necrotic cells in response to APAP by flow cytometry. The results shown in Figure 6 and Table 1 indicate a protection against both types of cell death in response to APAP in PTP1B-/- hepatocytes as compared to wild-type cells.
Effect of APAP treatment in the signaling pathways that modulate cell death and survival in wild-type and PTP1B-deficient hepatocytes.
At the molecular level, we analyzed both stress-mediated and survival signaling in wild-type and PTP1B-/- hepatocytes upon APAP treatment. The c-Jun N-terminal Kinases (JNKs), a subfamily of the mitogen-activated protein (MAP) kinases, have been shown to be activated by phosphorylation at early time-periods after APAP treatment in hepatocytes (38-39). To analyze JNK activation in immortalized hepatocytes, cells were treated with different doses of APAP for 8 h and JNK phosphorylation was examined by Western blot analysis. As shown in Figure 7A, APAP markedly increased the levels of phosphorylated JNK in wild-type hepatocytes. Interestingly, the lack of PTP1B prevented JNK activation in response to APAP.
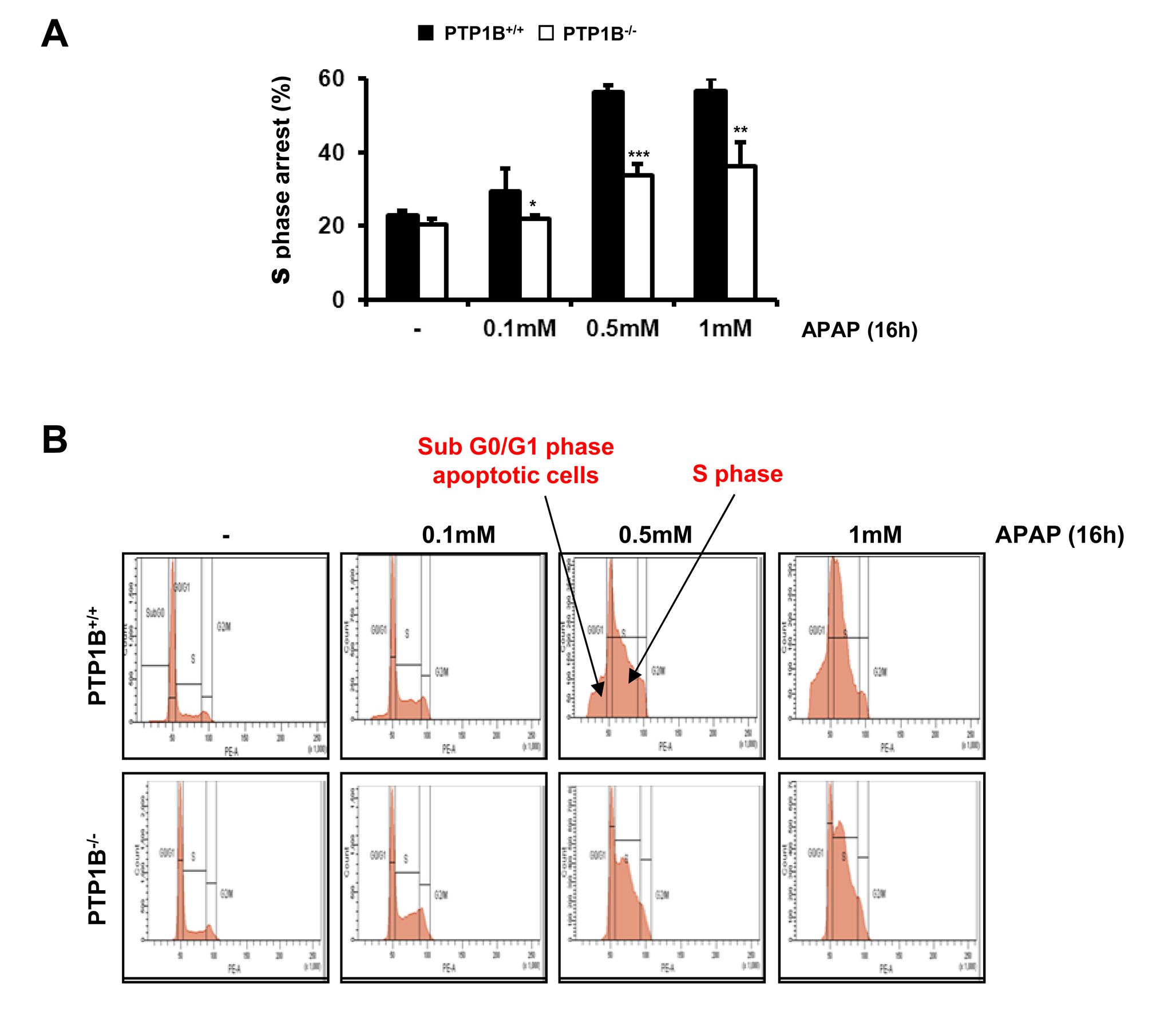
Figure 5.- PTP1B-deficient hepatocytes are protected against APAP-induced arrest in the S phase of the cell cycle. A. PTP1B+/+ and PTP1B-/- immortalized hepatocytes were treated with various doses of APAP for 16 h. The percentage of S phase cell population was measured by flow cytometry. B. Images of flow cytomerty showing the effect of APAP treatment on G0/G1 and S phases in both PTP1B+/+ and PTP1B-/- immortalized hepatocytes. *P<0.05, **P<0.01 and ***P<0.005 PTP1B-/- vs. PTP1B+/+ cells (n=4 independent experiments).
Table 1.- Effect of APAP treatment in the induction of both apoptosis and necrosis in PTP1B+/+ PTP1B-/- hepatocytes.
Population |
PI |
PI+* |
PI+dim** |
PTP1B+/+ C |
99.5 |
10 |
10.8 |
PTP1B+/+ 0.5mM |
98.5 |
22.7 |
24.1 |
PTP1B+/+ 1mM |
98.7 |
23.1 |
24.6 |
PTP1B-/- C |
99.9 |
4.5 |
3.9 |
PTP1B-/- 0.5mM |
99.5 |
7.2 |
5.7 |
PTP1B-/- 1mM |
99 |
10.2 |
6.7 |
PI*=Necrosis. PI**=Apoptosis
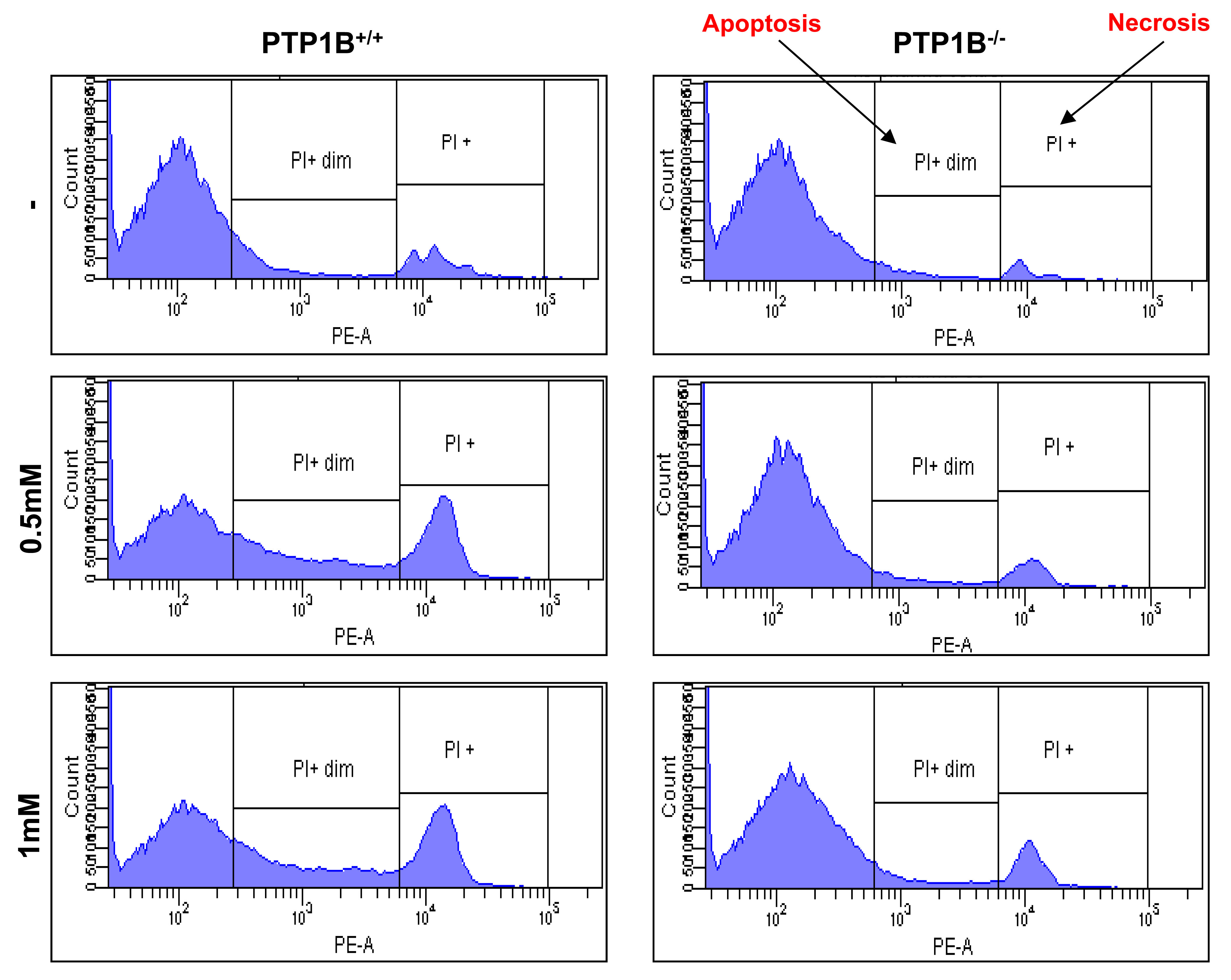
Figure 6.- PTP1B-deficient hepatocytes are protected against APAP-induced apoptosis and necrosis. PTP1B+/+ and PTP1B-/- immortalized hepatocytes were treated with various doses of APAP for 16 h. Apoptosis and necrosis was measured by PI staining by flow cytometry (n=4 independent experiments).
To study the effect of APAP on the survival signaling pathways, phosphorylation of the IGF-IR, levels of IRS1 and activation of Akt were examined. As depicted in Figure 7B, phosphorylation of the IGF-IR and its downstream target Akt decreased in APAP-treated wild-type hepatocytes at 8 h, but it was maintained along APAP treatment in PTP1B-deficient hepatocytes. These results indicate that the protective effect of PTP1B deficiency also involve the increase in survival signaling. Interestingly, in PTP1B-deficient hepatocytes IRS1 degradation induced by APAP was attenuated. Alltogether these results suggest that the increased tyrosine phosphorylation of IGF-IR as a result of PTP1B deficiency elicits hepatoprotection in conjunction with the attenuation of stress-mediated signaling.
Bcl-xL is a member of the Bcl-2 family with anti-apoptotic properties (40-41). On that basis, we analyzed the expression of Bcl-xL after 8 h of APAP treatment in wild-type and PTP1B-/- hepatocytes. As shown in Figure 7C, wild-type cells showed a decrease in the expression of BclxL after APAP treatment whereas BCLxL expression was maintained in hepatocytes lacking PTP1B.

Figure 7.- Effect of PTP1B deficiency in stress and survival signaling in hepatocytes. PTP1B+/+ and PTP1B-/- immortalized hepatocytes were treated with various doses of APAP for 8 h. Total cell lysates were analyzed by Western blot with the antibodies against phospho-JNK1/2 and JNK1/2 (A), phosphor-IGFIR, IGFIR, IRS1, phospho-Akt and Akt (B) and BclxL and β-actin as a loading control (C). Representative autoradiograms corresponding to three independent experiments are shown. D. PTP1B-/- immortalized hepatocytes were reconstituted with PTP1B by retroviral gene transfer. PTP1B-/- and PTP1B-/-Rec cells were treated with various doses of APAP for 16 h. Total cell lysates were analyzed by Western blot with the indicated antibodies. Representative autoradiograms corresponding to three independent experiments are shown.
Our next step was to study the effect of PTP1B reconstitution of PTP1B-deficient hepatocytes in the signaling pathways modulated by APAP. For this goal, we reconstituted PTP1B expression in deficient cells by retroviral gene transfer as described in Materials and Methods. As a control, hepatocytes were infected with an empty retroviral construct. Figure 7D shows the re-expression of PTP1B in PTP1B-/- immortalized neonatal hepatocytes (refrerred as PTP1B-/- Rec). Importantly, re-expression of recombinant PTP1B in PTP1B-/- neonatal hepatocytes induced a decline of IGF-IR phosphorylation after APAP treatment similar to the effects observed in wild-type cells. In addition, APAP treatment activated JNK phosporylation in PTP1B-/- Rec neonatal hepatocytes as in wild-type cells.
We have recently reported that APAP preotected against liver injury induced by a toxic dose of APAP (300 mg/kg) in mice (33). To reinforce this finding, we injected both PTP1B+/+ and PTP1B-/- mice with a higher dose of APAP (500 mg/kg) and analyzed liver toxicity. Serum ALT was higher in APAP-injected wild-type than in PTP1B-/- mice (Figure 8A). Moreover, less injury was observed in liver sections from APAP-injected PTP1B-/- mice as compared to the wild-type controls that presented histological signs of severe hepatotoxicity (Figure 8B).
PTP1B-deficient mice are protected against liver damage induced at a high dose of APAP.
We next determined if reduced sensitivity of PTP1B-/- mice to APAP-induced liver injury was associated with alterations in basal GSH levels in the two mose strains. Interestingly, the basal expression of Cyp2e1, GCL-C and GCL-M and basal GSH levels were comparable among both genotypes of mice (Figure 8C, 8D).

Figure 8.- PTP1B-deficient mice are protected against liver damage induced by a high dose of APAP under similar GSH levels and expression of Cyp2e1, GCL-C, GCL-M and BclxL in the liver. PTP1B+/+and PTP1B-/- mice were injected with 500 mg/kg APAP or saline for 6 h. A. ALT activity. **P<0.01 PTP1B-/- vs. PTP1B+/+ mice (n= 6-8 mice of each condition). B. Representative Hematoxylin & Eosin staining in liver sections. Bar scale 100 µm. C. Basal GSH levels in livers from PTP1B+/+ and PTP1B-/-mice. D. Whole cell lysates from both mice +were analyzed by Western blot with the indicated antibodies. Representative autoradiograms are shown.
4. conclusion
In summary, the present study provides the first molecular evidence that levels of PTB1B modulate susceptibility to apoptosis that occurred at early time-periods in hepatocytes treated with toxic doses of APAP. Thus, our results have revealed that PTP1B inhibition might be therapeutically of interest against the hepatotoxicity induced by overdoses of APAP.
5. Acknowledgements
We acknowledge the following grant support: SAF2012-33283 (MINECO, Spain), Comunidad de Madrid S2010/BMD-2423, EFSD and Amylin Paul Langerhans Grant and Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM, ISCIII, Spain).
6. references
1. Rumack, B. H. Acetaminophen misconceptions. Hepatology 40,10-15 (2004).
2. Thomas, S. H. L. Paracetamol (acetaminophen) poisoning. Pharmacol. Ther. 60, 91–120 (1993).
3. Litovitz, T. L.; Klein-Schwartz, W.; Rodgers, G. C.; Cobaugh, D. J.; Youniss, J.; Omslaer, J. C.; May, M. E.; Woolf, A. D.; Benson, B. E. 2001 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am. J. Emerg. Med. 20, 391–452 (2002).
4. Lee, W. M. Acetaminophen toxicity: changing perceptions on a social /medical issue. Hepatology 46, 966-970 (2007).
5. Kaplowitz, N. Acetaminophen hepatoxicity: what do we know, what don't we know, and what do we do next? Hepatology 40, 3-26 (2004).
6. Prescott, L. F. Hepatotoxicity of mild analgesics. Br. J. Clin. Pharmacol. 2, 373-379 (1980).
7. Lee, W. M. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology 40, 6–9 (2004).
8. Cohen, S. D.; Khairallah, E.A. Selective protein arylation and acetaminophen induced hepatotoxicity. Drug Metab. Rev. 29, 59-77 (1997).
9. James, L. P.; Mayeux, P. R.; Hinson, J. A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 31, 1499–1506 (2003).
10. Jaeschke, H.; Bajt, M. L. Intracellular signaling mechanisms of acetaminophen induced liver cell death. Toxicol. Sci. 89, 31–41 (2006).
11. Bessems, J. G,; Vermeulen, N. P. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit. Rev. Toxicol. 31, 55–138 (2001).
12. Nelson, S. G.; Wan, Z.; Stan, M. A. S (N)2 ring opening of beta-lactones: an alternative to catalytic asymmetric conjugate additions. J. Org. Chem. 67, 4680–4683 (2002).
13. Vermeulen N. P. E.; Bessems J. G. M.; Vandestraat R. Molecular aspects of paracetamol-induced hepatotoxicity and its mechanism-based prevention. Drug Metab. Rev. 24, 367–407 (1992).
14. Dong, H.; Haining, R. L.; Thummel, K. E.; Rettie, A. E.; Nelson, S. D. Involvement of human cytochrome P450 2D6 in the bioactivation of acetaminophen. Drug Metab. Dispos. 28, 1397-1400 (2000).
15. Snawder, J. E.; Roe, A. L.; Benson, R. W.; Roberts, D. W. Loss of CYP2E1 and CYP1A2 activity as a function of acetaminophen dose: relation to toxicity. Biochem. Biophys. Res. Commun. 203, 532-539 (1994).
16. Ghosh, A.; Sil, P. C. Protection of acetaminophen induced mitochondrial dysfunctions and hepatic necrosis via Akt-NF-KB pathway: role of a novel plant protein. Chem. Biol. Interact. 177, 96-106 (2009).
17. Mitchell, J. R,; Jollow, D. J.; Potter, W. Z.; Gillette, J. R.; Brodie, B. B. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 187, 211-217 (1973).
18. Reid, A.; Kurten, R.; McCullough, S.; Brock, R.; Hinson, J. Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J. Pharmacol.Exp. Ther. 312, 509–516 (2005).
19. Tonks, N. K.; Neel, B. G. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 13,182-195 (2001).
20. Frangioni, J. V.; Beahm, P. H.; Shifrin, V.; Jost, C. A.; Neel, B. G. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell 68, 545-560 (1992).
21. Flint, A. J.; Tiganis, T.; Barford, D.; Tonks, N. K. Development of "substrate-trapping" mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. 94, 1680-1685 (1997).
22. Haj, F. G.; Markova, B.; Klaman, L. D.; Bohmer, F. D.; Neel, B. G. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B. J. Biol. Chem. 278, 739-744 (2003).
23. Salmeen, A.; Andersen, J. N.; Myers, M. P.; Tonks, N. K.; Barford, D. Molecular basis for recognition and dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol. Cell 6, 1401-1412 (2000).
24. Buckley, D. A.; Cheng, A.; Kiely, P. A.; Tremblay, M. L.; O'Connor, R. Regulation of insulin-like growth factor type I (IGF-I)receptor kinase activity by protein tyrosine phosphatase 1B (PTP-1B) and enhanced IGF-I-mediated suppression of apoptosis and motility in PTP-1B-deficient fibroblasts. Mol. Cell Biol. 22, 1998-2010 (2002).
25. Fabregat, I.; Herrera, B.; Fernández, M.; Alvarez, A. M.; Sánchez, A.; Roncero, C.; Ventura, J. J.; Valverde, A. M.; Benito, M. Epidermal growth factor impairs the cytochrome C/caspase-3 apoptotic pathway induced by transforming growth factor beta in rat fetal hepatocytes via a phosphoinositide 3-kinase-dependent pathway. Hepatology 32, 528-35 (2000).
26. Valverde, A. M.; Fabregat, I.; Burks, D. J.; White, M. F.; Benito, M. IRS-2 mediates the anti-apoptotic effect of insulin in neonatal hepatocytes. Hepatology 40, 1285–1294 (2004).
27. Gonzalez-Rodriguez, A.; Clampit, J. E.; Escribano, O.; Benito, M.; Rondinone, C. M.; Valverde, A. M. Developmental switch from prolonged insulin action to increased insulin sensitivity in protein tyrosine phosphatase 1B-deficient hepatocytes. Endocrinology 148, 594– 608 (2007).
28. Gonzalez, R. A.; Gutierrez, J. A.; Sanz-Gonzalez, S.; Ros, M.; Burks, D. J.; Valverde, A. M. Inhibition of PTP1B restores IRS1-mediated hepatic insulin signaling in IRS2-deficient mice. Diabetes 59, 588-599 (2010).
29. Gonzalez-Rodriguez, A.; Mas-Gutierrez, J. A.; Mirasierra, M.; Fernandez-Perez, A.; Lee , Y. J.; Ko, H. J.; Kim, J. K.; Romanos, E.; Carrascosa, J. M.; Ros, M.; Vallejo, M.; Rondinone, C. M.; Valverde, A. M. Essential role of protein tyrosine phosphatase 1B in obesity-induced inflammation and peripheral insulin resistance during aging. Aging Cell 11, 284-296 (2012).
30. Granado-Serrano, A. B.; Martín, M. A.; Goya, L.; Bravo, L.; Ramos, S. Time-course regulation of survival pathways by epicatechin on HepG2 cells. J. Nutr. Biochem. 20, 115-124 (2009).
31. Alía, M.; Ramos, S.; Mateos, R.; Bravo, L.; Goya, L. Response of the antioxidant defense system to t-butyl hydroperoxide and hydrogen peroxide in a human hepatoma cell line (HepG2). J. Biochem. Mol. Toxicol. 19, 119–28 (2005).
32. Schulte-Hermann, R.; Bursch, W.; Grasl-Kraupp, B. Active cell death (apoptosis) in liver biology and disease. Progress Liver Disease 13, 1-35 (1995).
33. Mobasher, M. A.; Gonzalez-Rodriguez, A.; Santamaria, B.; Ramos, S.; Martin, M. A.; Goya, L.; Rada, P.; Letzig, L.; James, L. P.; Cuadrado, A.; Martın-Perez, J.; Simpson, K. J.; Muntane, J.; Valverde, A. M. Protein tyrosine phosphatase 1B modulates GSK3beta/Nrf2 and IGFIR signaling pathways in acetaminophen-induced hepatotoxicity. Cell Death Dis. 4: e626 (2013).
34. Sangwan, V.; Paliouras, G. N.; Cheng, A.; Dube, N.; Tremblay, M. L.;Park, M. Protein-tyrosine phosphatase 1B deficiency protects against Fas-induced hepatic failure. J. Biol. Chem. 281, 221–228 (2006).
35. Kon, K.; Kim, J. S.; Jaeschke, H.; Lemasters, J. J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 40, 1170–1179 (2004).
36. Ferret, P. J.; Hammoud, R.; Tulliez, M.; Tran, A., Trébéden, H.; Jaffray, P.; Malassagne, B.; Calmus, Y.; Weill, B.; Batteux, F. Detoxification of reactive oxygen species by a nonpeptidyl mimic of superoxide dismutase cures acetaminophen-induced acute liver failure in the mouse. Hepatology 33, 1173–1180 (2001).
37. Boyce, M.; Degterev, A.; Yuan, J. Caspases: an ancient cellular sword of Damocles. Cell Death Differ. 11, 29-37 (2004).
38. Gunawan, B.; Liu, Z. X.; Han, D.; Hanawa, N.; Gaarde, W. A.; , Kaplowitz, N. c-Jun N terminal kinase plays a major role inmurine acetaminophen hepatotoxicity. Gastroenterology 131,165–178 (2006).
39. Bae, M.; Pie, J.; Song, B. Acetaminophen induces apoptosis of C6 glioma cells by activating the c-Jun NH2-terminal protein kinase-related cell death pathway. Mol. Pharmacol. 60, 847–856 (2001).
40. Chao, D. T.; Korsmeyer, S. J. BCL-2 family: regulators of cell death. Annu. Rev. Immunol. 16, 395–419 (1998).
41. Green, D. R.; Reed, J. C. Mitochondria and apoptosis. Science 281, 1309–1312 (1998).