REVISIÓN |
Envejecimiento cerebral normal y patológico: continuum fisiopatológico o dualidad de procesos involutivos
Adolfo Toledano 1,2,*, María-Isabel Álvarez1, Adolfo Toledano-Díaz3
1Instituto Cajal, CSIC, Madrid. 2Académico Correspondiente de la Real Academia Nacional de Farmacia. 3Departamento de Reproducción, INIA, Madrid
*e-mail: atoledano@csic.cajal.es
Recibido el 7 de abril de 2014 An. Real Acad. Farm. Vol. 80, Nº 3 (2014), pag. 500-539
resumen
Desde hace años existe una controversia sobre si existe una doble vía (dualidad involutiva) de involución senil del cerebro, que podemos denominar, respectivamente, hacia la senilidad fisiológica o “normal” sin signos de demencia, o bien hacia la senilidad “patológica” o Enfermedad de Alzheimer, que se manifiesta por la aparición de una demencia progresiva, o, por el contrario, una sola vía (continuum) que conduce desde los primeros signos de involución morfológica o funcional senil hasta la fase final de la demencia EA en todos los individuos. Además, en estos últimos años, se está produciendo una profunda revisión de los conceptos sobre la senilidad patológica / EA de manera que ya no se considera que dicho proceso de envejecimiento sólo existe tras efectuar un diagnóstico de demencia, sino que se inicia en el momento en que se produce una alteración de los circuitos cognitivos cerebrales y/o cambio neuropatológico. Con estos nuevos criterios se supone que la senilidad patológica / EA comprende diversas fases sin demencia (asintomáticas o prodrómicas) y otras con demencia de distinto grado. En esta revisión se analizan las razones que se aducen en defensa de una u otra teoría y las características morfofuncionales de los cerebros seniles normales y los patológicos. La mayoría de los estudios realizados apuntan que existen características diferenciales muy marcadas entre los cerebros seniles normales sin demencia y los patológicos con demencia, pero en otros existen grandes discrepancias entre la presencia o no demencia y la de signos de neuropatológicos. La existencia o no del continuum tiene también una muy importante repercusión práctica pues tanto la prevención como la asistencia de las personas mayores dependerán del posible número de individuos afectados: bien solamente los individuos de riesgo o bien toda la población. Existen algunos terrenos donde la investigación podría hacer avanzar nuestros conocimientos en el envejecimiento cerebral, especialmente en el estudio pormenorizado de los individuos controles sanos de entre 30 y 60 años, en el de los “centenarios” con y sin demencia y en grupos con características diferenciales neuropsicológicas (posibles estadíos intermedios de EA según el nuevo “lexicón”). También sería de gran interés estudiar comparativamente el envejecimiento cerebral humano con el de otras especies de mamíferos que no presentaran patología amiloide o tau y con los primates que pueden presentar patología amiloidea. En este último sentido, los animales transgénicos con patología inducida amiloide o/y tau también serían importantes fuentes de información. |
Palabras clave: Senilidad cerebral normal; senilidad cerebral patológica; continuum; Enfermedad de Alzheimer.
abstract
Normal and pathological brain aging: a physiopathological continuum or an involutive duality
For years, there is a controversy whether there is a two-way (involutive duality) of senile involution of the brain, which may be called, respectively, pathway to the physiological or " normal" senility, without signs of dementia but with neuronal adaptative responses, and pathway to "pathological " senility or Alzheimer's Disease (AD), which is characterized by the appearance of a progressive dementia, or, conversely, a single way (continuum) leading from the first morphological or functional signs of senile involution until the final stage of AD dementia in all individuals. Moreover, in recent years, a thorough review of the concepts of the pathological senility / EA is producing, so that is not now considered necessary to have dementia to diagnose a pathological aging process because it is considered that the pathological aging / EA is started at the time that a disturbance in cognitive brain circuits and / or neuropathological changes occurs. With these new criteria different clinic-pathological stages are supposed from normal senility to terminal pathological senility / EA, without dementia (prodromal or asymptomatic) and with varying degrees of dementia. In this review the reasons given in defense of one or another theory are analyzed, as well as the morphofunctional features of normal and pathological senile brains. Most studies indicate that there are very marked differential characteristics between normal brains without dementia and senile pathological brains with dementia, but in other, large discrepancies exist between the presence or absence of dementia and the existence or not of neuropathological signs. The existence or not of the continuum also has a very important practical implication for both prevention and care of the elderly, because the possible number of individuals affected: the entire population or only at-risk individuals in the pathological brain senility pathway. There are some areas where research could improve our knowledge on brain aging, especially in the detailed study of healthy control subjects between 30 and 60, “centenarians” with and without dementia and individuals with neuropsychological characteristics different to AD (possible intermediate stages of AD according to the new " lexicon "). It would also be of great interest a comparative study of human brain aging with that of other mammalian species that showed no amyloid or tau pathology and primates that may have amyloid pathology. Transgenic animals with induced amyloid and /or tau pathology, are also important sources of information on pathological aging. |
Keywords: Normal brain senility; pathological brain senility; continuum; Alzheimer´s Disease.
1. INTRODUCcióN
La población mundial crece a un ritmo del 1% anual, con diferencias notables entre los países según su grado de desarrollo y los tramos de edad en que normalmente se dividen las pirámides poblacionales (1). En los países más desarrollados el porcentaje de mayores de 65 años aumenta en mayor grado al no existir recambio poblacional (1, 2). En países como España, las personas mayores de 65 años representan ya el 18% de la población (1). Paralelamente, los recursos y los fondos que los estados dedican a este sector de población son cuantiosos y también tienden a crecer. Todo ello genera una gran preocupación y las políticas socio-sanitarias pasan a ser prioritarias en la gestión de los gobiernos. En este contexto, las patologías relacionadas con la edad se consideran de especial gravedad porque, junto al sufrimiento de los que las padecen y de sus familiares, significan que un número importante de personas precisa mayores atenciones sanitarias y sociales, tanto más cuando se es sujeto de una discapacidad. Los trastornos mentales que cursan con demencia, y especialmente el más paradigmático, la enfermedad de Alzheimer (EA), por las características de la discapacidad que acarrea, de que se trata de una patología que actualmente no tiene prevención ni tratamiento eficaz, y en la que el enfermo requiere la continua presencia de un cuidador, son uno de los mayores problemas del siglo XXI. En el mundo, en 2010, se estimaba que de 758,6 millones de personas mayores de 60 años, existían más de 35,6 millones de personas con demencia (4,7% del total). De ellas, más del 80% eran enfermos de EA (2), que pueden considerarse personas con envejecimiento cerebral “patológico” no dependiente de ninguna enfermedad sistémica o cerebral conocida frente a los ancianos sin trastornos mentales que tienen un envejecimiento cerebral fisiológico/”normal”. Y las previsiones para 2.030 y 2.050 calculaban cifras de 65,7 y 115,4 millones de enfermos con demencia (incrementos del 85 y 225% de los valores de 2010), con porcentajes similares de aumento en EA. Los incrementos a partir de 2025 se han corregido al alza teniendo en cuenta que los “baby boomers”, los niños nacidos en gran número tras el fin de la segunda guerra mundial, cumplen 80 años en esa fecha (3, 4). En 2010, en España existían 0,4 millones de enfermos EA y se preveían 0,7 millones en 2030 y 1,6 millones en 2050, agravándose el problema socio-sanitario y económico español por la drástica disminución de la población más joven que debería actuar tanto como “cuidador” directo del enfermo EA como de “sostén”/”financiador” del sistema socio-sanitario.
Para enfrentarse a los problemas que esta situación plantea, trabajan con especial empeño la medicina preventiva y la planificación socio-sanitaria. Pero para actuar lo más eficazmente en estos dos ámbitos es absolutamente necesario conocer con la mayor exactitud posible los casos que realmente deberán ser atendidos en una época determinada, tanto en los aspectos preventivos como paliativos. Los resultados de los estudios en diversos campos de los últimos años sobre el envejecimiento cerebral humano y las patologías demenciales concurrentes obligan a revisar nuestras previsiones sobre la población de mayores de 60 años que ha aumentado enormemente su límite de supervivencia. No sólo debemos basar las actuaciones en los datos epidemiológicos sobre las enfermedades demenciales, sino también en los nuevos hallazgos sobre la patogenia de la EA y otras demencias y en las posibilidades terapéuticas para combatirlas, tanto preventiva como curativamente. En lo que respeta a la EA, muchos de los nuevos conceptos sobre la enfermedad (5, 6) han puesto de manifiesto la posible existencia de diferentes fases, entidades patológicas y/o subtipos de EA (EA típicas y EA atípicas) que pueden imbricarse de distintas formas, bien como fases consecutivas de un proceso continuado de mayor entidad que lleva a la EA terminal, o bien como formas involutivas o patologías alternativas en la degeneración senil del cerebro (7-9). Así mismo, la diversidad y complejidad de las alteraciones neuropatológicas y neuroquímicas que se van describiendo nos proporcionan un mosaico de posibilidades de nuevos diagnósticos de entidades todavía no bien definidas pero que expresan la posible existencia de diversos cursos patogénicos, o “cascadas” patogénicas, que se pueden imbricar en algunos puntos de la involución, y que sólo tienen en común el deterioro final total de los circuitos cognoscitivos cerebrales que subyace en la demencia terminal (10-12).
En la presente revisión se va a tratar una de las cuestiones de mayor interés en las demencias tipo EA: ¿existe un envejecimiento cerebral normal sin demencia y otro patológico con demencia tipo EA?, o por lo contrario, ¿existe sólo un envejecimiento progresivo donde de manera continuada se pasa por fases seniles sin demencia a otras con alteraciones cognoscitivas leves y que finaliza en lo que ahora consideramos la EA terminal?. Aceptar uno u otro postulado supone un gran cambio de estrategia tanto en la política sanitaria como en medicina preventiva. En el caso de España, si consideramos cierta la dualidad del curso involutivo, sin demencia o con demencia, habría que incluir en los cálculos asistenciales “solamente” el porcentaje de población susceptible de padecer EA (1,6 millones de personas en 2050), pero si se considera que existe un continuum, se deberían establecer medidas preventivas y sistemas asistenciales para más de 13,5 millones de españoles que se verán afectados hacia mitad del siglo XXI (1,13). Por otro lado, si los nuevos postulados sobre la diversidad de procesos patogénicos incluidos en el universo Alzheimer (2, 5, 6, 13) se confirman, el número de afectados debería revisarse al alza aunque habría que considerar como elemento corrector la posible eficacia de nuevas medidas preventivas específicas que actúen sobre las nuevas dianas terapéuticas que va ofreciendo la investigación. Además, habría que intensificar las medidas preventivas en individuos asintomáticos en edad adulta (40-50años) para combatir las alteraciones patológicas más precoces que pudieran aparecer en el inicio del envejecimiento. Actualmente es un reto el diseño de métodos y herramientas para definir la posible población en riesgo para ser especialmente tratada contra la neurodegeneración.
La polémica sentencia “todos padeceremos Alzheimer con tal de que vivamos el suficiente número de años” ha trascendido ya fuera de los ámbitos científicos y tiene unas implicaciones socio-sanitarias y económicas importantísimas que sólo la investigación puede fijar en los justos límites al definir y diagnosticar con precisión los individuos en riesgo de padecer EA. En esta revisión se analizará tanto el concepto de continuum entre el envejecimiento cerebral normal y patológico dentro de las nuevas teorías sobre la etiopatogenia y clínica de la EA y los argumentos científicos que sustentan o niegan su existencia, como las líneas de investigación actuales más importantes para esclarecer este importante problema.
2. Involución cerebral senil normal y patológica. La Enfermedad de Alzheimer
El ciclo vital nacimiento-desarrollo-envejecimiento-muerte se cumple en todos los seres vivos. Todos los órganos y sistemas de los seres del reino animal también cumplen este ciclo desde la formación de su primordio hasta el fallecimiento del ser que los alberga, aunque las características de este proceso de desarrollo e involución sean muy diferentes ya que son específicas de cada uno de ellos.
El Sistema Nervioso Central (SNC) presenta un desarrollo de diseño muy especial (14, 15). En el período embrionario de cada especie, se crea una ingente cantidad de neuronas pero sólo sobreviven las que emigran hacia lugares predeterminados, los “núcleos grises” del SNC, y establecen conexiones correctas con otras neuronas del propio núcleo al que pertenecen o de otros núcleos más o menos alejados, que también están predeterminados para cada especie. Se crea así la citoarquitectonia básica del SNC, fundamento de los circuitos neuronales, que se presenta al nacimiento del ser. En la época postnatal, salvo contadas excepciones en que permanecen áreas de neurogénesis (p.e., en mamíferos, el cerebelo –capa de Obersteiner, 16- y los centros germinales del hipocampo y de la zona paraventricular -17) el desarrollo se centra en el establecimiento y refuerzo de nuevas conexiones neuronales que funcionarán de manera dinámica para cumplir todas las funciones encomendadas al SNC, desde las sencillas respuesta monosinápticas que son la base de algunos movimientos reflejos, hasta las muy complejas respuestas multisinápticas donde intervienen millones de neuronas para cumplir las funciones cognoscitivas superiores (los diversas tipos de memoria, el juicio, etc.). Además, se ha demostrado que las sinapsis son “plásticas” (18-21), es decir cambian morfológicamente en número, forma y tamaño, así como funcionalmente, aumentando o disminuyendo los componentes macromoleculares específicos para su función y regulando la producción de neurotransmisores, neurorreguladores y segundos mensajeros intracelulares. Esta plasticidad sináptica no es más que una manifestación de la gran capacidad de adaptación que tiene las neuronas (en realidad, son las células que poseen la mayor capacidad de adaptación morfofuncional de todo el organismo) para que sus respuestas sean siempre óptimas y cumplan con los objetivos que cada parte del SNC tiene encomendados (21). Esta adaptación ocurre a lo largo de toda la vida incluso en la fase senil: el envejecimiento cerebral debe entenderse como un proceso involutivo –adaptativo (22) donde no existe reemplazamiento de neuronas, por ser células postmitóticas incapaces de dividirse, pero sí de funciones de algunas neuronas para paliar en parte las pérdidas funcionales debidas a las neuronas desaparecidas o dañadas. Neuronas sobrevivientes que funcionaban en paralelo a neuronas perdidas dentro de diversos circuitos cognoscitivos, pueden suplantar la función de estas últimas por lo que durante mucho tiempo no se alterará la función cognoscitiva relacionada con el circuito afectado y tienen que ser muchas las neuronas deterioradas o desaparecidas para que la función cognoscitiva de muestras de deterioro. Se ha comprobado de manera experimental como aumentan las sinapsis de muchas regiones del SNC de diversos animales seniles con entrenamiento o bien con su permanencia en “ambientes enriquecidos” (jaulas con juguetes o laberintos) (22, 23). También pensemos que nuestra especie es capaz de leer, entender y aprender incluso al borde del fin de nuestra existencia, pues existen fenómenos adaptativos en las neuronas supervivientes, que incluyen neosinaptogénesis (24), para mantener muchas de las funciones cognoscitivas. Es decir, a partir de cierta edad en que se alcanza la madurez (entre los 30 y los 50 años), muchos de los circuitos neuronales establecidos y perfeccionados por las conexiones sinápticas desde el nacimiento se van a modificar, pero esto ocurre tanto por el proceso involutivo senil, donde mueren o disfuncionan muchas neuronas anulando o dañando los circuitos, como por la puesta en marcha de mecanismos adaptativos para contrarrestar estas pérdidas morfofuncionales (Figura 1).
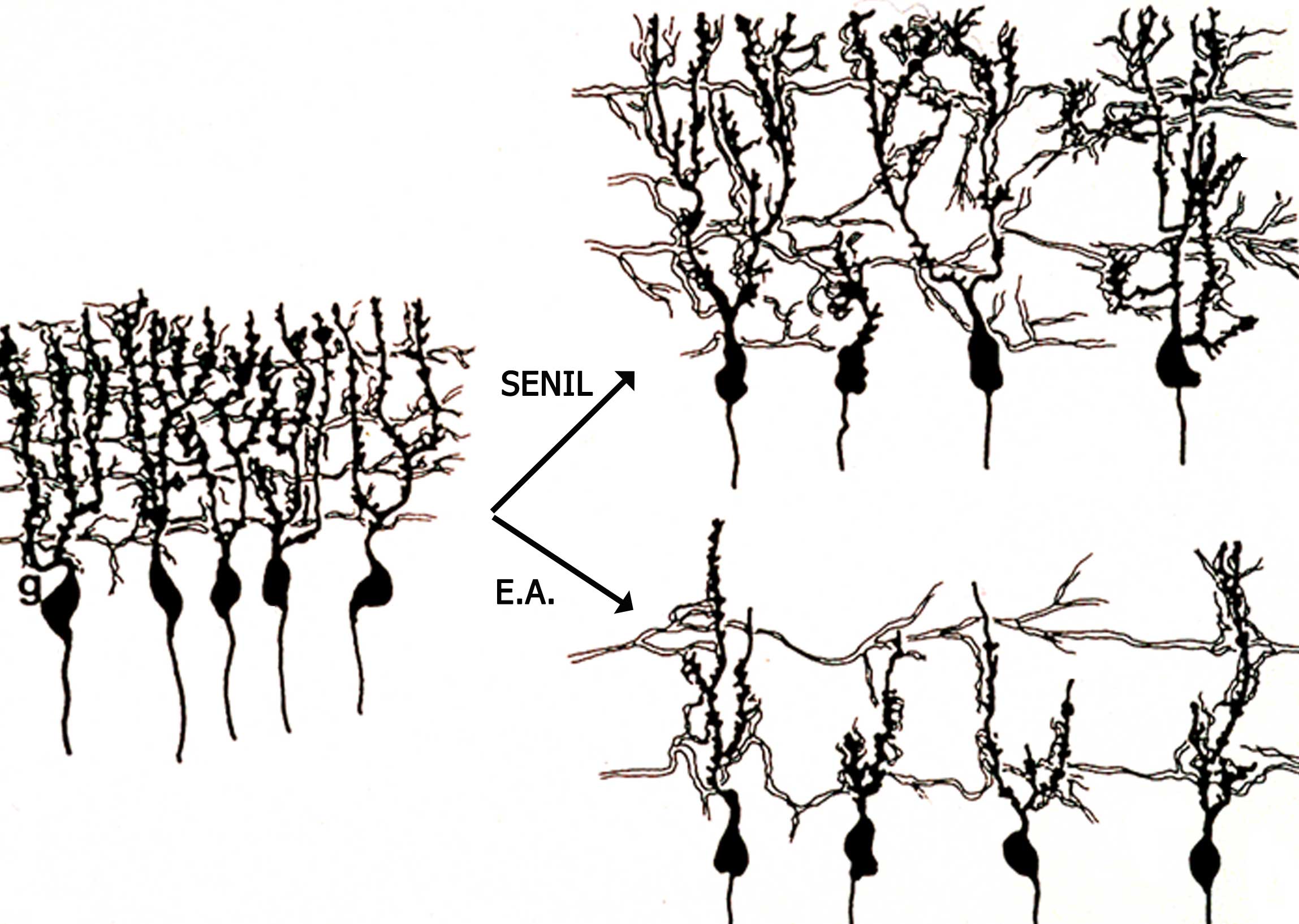
Figura 1.- Esquema que representa las diferencias fundamentales entre el proceso de “senilidad normal” (senil) y el de “senilidad patológica” en las células piramidales del hipocampo. En ambos casos existe pérdida neuronal y de fibras aferentes, pero en la senilidad normal o fisiológica, los procesos adaptativos originan crecimiento dendrítico neuronal y neosinaptogénesis en las neuronas remanentes y en las fibras aferentes a las mismas. Esto no sucede en la enfermedad de Alzheimer o senilidad patológica.
En un cerebro senil “normal” encontraremos un menor número de neuronas en diversas regiones del SNC (aunque no tan grande como se señalaba hace unos años), una más importante pérdida de sinapsis sobre muchas neuronas y una disminución de moléculas importantes para la conexión sináptica y la neurotransmisión. Se pueden apreciar neuronas atróficas y distróficas, la base del proceso de involución senil, pero también hipertróficas y/o hiperactivas debidas a la adaptación neuronal, con aumento de sinapsis recibidas y/o sinapsis producidas por sus axones o bien que posean una mayor capacidad de producción de neurotransmisores, mensajeros extracelulares, receptores o moléculas específicas de la función sináptica. En general, no parecen existir alteraciones estructurales muy importantes de neuronas ni tampoco de las células gliales acompañantes (astrogliales y microgliales) ni de los vasos sanguíneos. Los individuos cuyos cerebros tienen estas características, no presentaban alteraciones muy importantes o globales de las funciones cognoscitivas y comportamentales; es decir, no padecían demencia cuya característica, según la OMS (25), es “la pérdida de múltiples –todas- funciones cognoscitivas como son la memoria-s-, el pensamiento abstracto, el juicio, …”). Sólo en algunos casos se apreciaban ligeros deteriores en memoria, lenguaje, cálculo, etc.
Por el contrario, existen individuos seniles que manifiestan un grado variable de demencia (de leve a grave), con alteraciones importantes de todas las funciones cognoscitivas, aunque no todos los deterioros cognoscitivos se hayan iniciado simultáneamente ni su progresión curse en paralelo. El estudio del cerebro de estos individuos muestra que, además de las alteraciones antes mencionadas del cerebro senil “normal”, aparecen lesiones o cambios aberrantes en las neuronas y en las células gliales. No sólo se trata de una “exageración” del deterioro que consideramos normal sino que nos encontramos en presencia de nuevos elementos que consideramos patológicos (neuropatológicos) dentro y fuera de las células (“ovillos neurofibrilares” y “placas amiloides”, en especial). Hablamos entonces que ha existido una neurodegeneración o involución neurodegenerativa que se manifiesta clínicamente en vida con demencia. Se trata de un “envejecimiento senil patológico” por contraposición al anteriormente reseñado, el “envejecimiento senil normal o fisiológico”.
Alois Alzheimer, en 1906-7 (26), fue el primero en describir en el cerebro de una mujer que había padecido demencia, una serie de alteraciones neuropatológicas (luego denominados ovillos y placas) que aunque inicialmente consideró como signos de senilidad precoz, luego describió como signos de una enfermedad nueva o senilidad patológica frente la senilidad “normal de la mayoría de los individuos”. Esto fue así confirmado por Kraepellin en 1910 (27) y posteriormente avalado por la investigación científica básica y clínica. A lo largo de los años han ido evolucionando muchos de los conceptos sobre la Enfermedad de Alzheimer, pero esta idea de que se trata de un envejecimiento cerebral patológico, permanece inalterable.
De la imposibilidad de estudiar en profundidad y con los mismos métodos el cerebro de los humanos vivos y el de los ya fallecidos, tanto de individuos con senilidad normal como patológica (individuos que habían padecido demencia), surge el concepto clásico dual de la enfermedad, clínico en vida y neuropatológico post-mortem. Estos conceptos “clásicos” (de la época de Alzheimer) se mantienen durante muchos años y son la base de las descripciones “modernas” de la enfermedad tales como se recogen en la CIE-10 de la OMS (1960) (25) y en el manual de Diagnóstico Estadístico (DMS-III, DMS-IV y DMS-V) de la Academia de Patología Americana (1960, 1970 y 2014) (28, 29), donde se marcan pautas para el diagnóstico. Diversos estudios de consenso establecen protocolos de diagnóstico, tanto pruebas neuropsicológicas desarrolladas exprofeso para caracterizar con mayor precisión la demencia en la clínica, como estudios anatomopatológicos pormenorizados para tipificar y cuantificar las lesiones de los cerebros post-mortem (30, 31). El diagnóstico en vivo se lleva a cabo cuando existe un cuadro demencial de comienzo insidioso y deterioro lento con ausencia de datos clínicos sugerentes de enfermad cerebral o sistémica que den razón de la demencia (Cuadro I) (25). Es decir, se realiza un diagnóstico de presunción por exclusión de otra posible patología, neurodegenerativa o no. La confirmación post-mortem del diagnóstico, también ante la ausencia de signos neuropatológicos patognomónicos, se hace de manera estadística, cuando los tipos y la abundancia de lesiones según un estudio estandarizado nos indica, con un alto índice de probabilidad, que existe ésta y no otra enfermedad (30, 31).
CUADRO I
GLOSARIO DE DEFINICIONES SOBRE SITUACIONES Y PROCESOS FISIOPATOLÓGICOS CEREBRALES Y COGNOSCITIVOS EN LA SENILIDAD Y PAUTAS DIAGNÓSTICAS GENERALES. SENILIDAD FISIOLÓGICA: Proceso en el que se presentan alteraciones leves en algunas áreas cognoscitivas (memorias, cálculo, juicio, etc., sin llegar a presentar un cuadro de demencia), comportamentales, sensitivas y motoras que no repercuten de manera importante en la vida del sujeto. Existen alteraciones morfofuncionales involutivas pero no neuropatología en sentido clásico. SENILIDAD PATOLÓGICA: Proceso en el que se presentan de manera progresiva alteraciones importantes de casi todas las áreas cognoscitivas, llegando a presentar un cuadro de demencia, con alteraciones graves comportamentales, sensitivas y motoras que repercuten de manera trascendente en la vida del sujeto, haciéndole totalmente dependiente. Existen alteraciones neuropatológicas en sentido clásico. Se asimila a Enfermedad de Alzheimer por la mayoría de autores. DEMENCIA (CIE-10) (25): “Síndrome debido a una enfermedad del cerebro, generalmente de naturaleza crónica y progresiva, en la que hay déficits de múltiples funciones corticales superiores, entre ellas la memoria, el pensamiento, la comprensión, el cálculo, la capacidad de aprendizaje, el lenguaje y el juicio” Interfiere en la realización de las actividades de la vida diaria del individuo hasta llegar a hacerlo totalmente dependiente ENFERMEDAD DE ALZHEIMER (SENILIDAD PATOLÓGICA). DEFINICIÓN CLÁSICA DE LA EA Y SU DIAGNÓSTICO (CIE-10) (25): -(Definición basada en estudios post-mortem del cerebro) “Enfermedad degenerativa cerebral primaria, de etiología desconocida que presenta rasgos neuropatológicos y neuroquímicos característicos” - (Definición dignóstica in vivo). Proceso caracterizado por: * Cuadro demencial * Comienzo insidioso y deterioro lento * Ausencia de datos clínicos sugerentes de enfermedad cerebral o sistémica * Ausencia de inicio apopléjico, súbito o con signos neuropatológicos focales Repercute de manera importante en la vida del sujeto, interfiriendo en su vida profesional y social y al no poder llevar a cabo las actividades de la vida diaria le hacen dependiente (DMS III-V tiene criterios diagnósticos similares aunque les conceda diferente valoración) (28, 29) NUEVAS DEFINICIONES DEL COMPLEJO EA Y SU DIAGNÓSTICO (Dubois y cols, 2007;2010) (5, 6). Enfermedad de Alzheimer Entidad clínico biológica que comprende varias fases sin y con síntomas de demencia hasta llegar a la situación que se corresponde con la EA clásica. Para su diagnóstico se requiere cambios cognoscitivos (memoria) y presencia de “marcadores” de EA (betamiloide, tau y fosfo-tau en LCR; retención de trazadores de amiloide en PET cerebral; atrofia temporal en RM cerebral; alteraciones metabólicas como captación de 18 Fluorodeoxiglucosa en PET; etc.). Pueden existir formas fenotípicamente típicas y atípicas de EA. Comprende la EA prodómica y la demencia EA. EA prodrómica (o estadio de “predemencia EA”) Fase temprana de la demencia EA que presenta: 1) síntomas de alteración cognoscitiva (especialmente pérdida de memoria de origen hipocámpico), pero sin entidad suficiente para diagnosticar demencia y sin interferir en la realización de actividades de la vida diaria y 2) alteraciones de los biomarcadores EA. Fase preclínica de la EA Largo período asintomático de la EA, teóricamente desde que se produce la primera lesión cerebral tipo Alzheimer en un individuo normal hasta que se presentan síntomas de alteración cognoscitiva. Comprende dos estadios: “Estadio Asintomático de la EA” o “ Estadio de alto riesgo para padecer EA”. Se podría identificar demostrando amiloidosis cerebral (p.e., mediante PET con el complejo Pittsburg) o alteraciones “EA” en LCR. “EA Presintomática”. Periodo previo a la EA sintomática con posibles signos de cambio. Sólo podría diagnosticarse con certeza en EAs familiares con mutaciones dominantes. Demencia EA Fase de la EA en la que los trastornos cognoscitivos en el área de la memoria y otras áreas son tan graves que interfieren con la vida social y laboral y la realización de las actividades de la vida diaria (existe un diagnóstico claro de demencia). EA típica El fenotipo más común de EA, caracterizado por pérdida progresiva de la memoria episódica, que es dominante, y a la que se asocian progresivamente pérdidas en otras áreas (ejecutivas, del lenguaje, visuales, etc). EAs atípicas Fenotipos clínicos poco corrientes, de un curso clínico anómalo comparado con la EA típica. Son entidades descritas en los últimos años, algunas mal definidas. Se pueden señalar: afasia primaria progresiva, afasia logopénica, EA frontal, atrofia cortical posterior. El diagnóstico de EA atípica precisa de la demostración de alteraciones en biomarcadores EA. EA mixta Concurrencia de EA típica con evidencias de neuroimagen o bioquímicas de otros procesos comórbidos como Enfermedad Cerebrovascular o Enfermedad de Cuerpos de Lewy. Patología Alzheimer. Individuos sin demencia con alteraciones neuropatológicas EA (depósitos de amiloide y tau). Alteración (o deterioro) cognoscitivo leve (MCI, “mild cognitive impairement”). Alteraciones cognoscitivas leves, que no interfieren en la vida diaria, sin causa aparente (se diagnostica también por exclusión). No presenta características clínicas para el diagnóstico de EA ni tiene alteraciones de los marcadores EA. Puede llegar a presentar EA en un plazo de 3-5 años. |
En los últimos años, los resultados de la investigación clínica, neurobioquímica celular y molecular, y neuropatológica, han venido a revolucionar los conceptos sobre la EA de tal manera que se considera la enfermedad como un proceso neurodegenerativo que se inicia mucho antes de que se pueda diagnosticar la demencia en el aspecto clínico, justo cuando se inician las lesiones de los circuitos neuronales (la denominada EA prodrómica) (Cuadro I) (5, 6). El problema reside en que tampoco es todavía posible detectar los cambios morfofuncionales cerebrales con la tecnología actual. Además, se considera que existen unas fases en las que sin existir toda vía la demencia, ya se pueden detectar alteraciones cognoscitivas previas a la demencia, especialmente el deterioro cognitivo leve (5-7).
También, como resultado de estas nuevas investigaciones de los últimos diez años, se han venido descubriendo nuevas entidades de envejecimiento cerebral que, aunque serían minoritarias frente a las dos principales, tienen un gran interés por su posible implicación en ellas. Por un lado parecen existir individuos que se sitúan en ámbitos clínicos entre la demencia y la inexistencia de demencia durante muchos años y los escasos estudios anatomopatológicos que se han realizado con sus cerebros post-morten han mostrado diversidad en el grado de anomalías EA, sin poderse afirmar si eran situaciones patológicas que podrían situarse en la fase prodrómica de la EA, o bien entidades “nuevas” distintas a la involución fisiológica normal y a la EA (32). Por otro lado, se han encontrado cerebros de dementes “clínicamente EA” con alteraciones neuropatológicas que no se corresponden con la patología típica de la EA (EA atípica) (Figura 2). Existen casos sin la típica presencia y distribución de placas y de ovillos neurofibrilares o con la presencia de otro tipo de alteraciones muy marcadas (33, 34). En este último caso, en los últimos años se ha impuesto la tendencia a segregar estas entidades en nuevas en enfermedades seniles que cursan con demencia que se situarían entre la enfermedad de Alzheimer (típica y atípicas) y otras neurodegeneraciones seniles no Alzheimer como la enfermedad de Creutzfeld-Jakob (priónica) o el Parkinson-demencia. La demencia frontotemporal, la demencia de cuerpos de Lewy y la atrofia hipocámpica son ejemplos de ello (33-36).
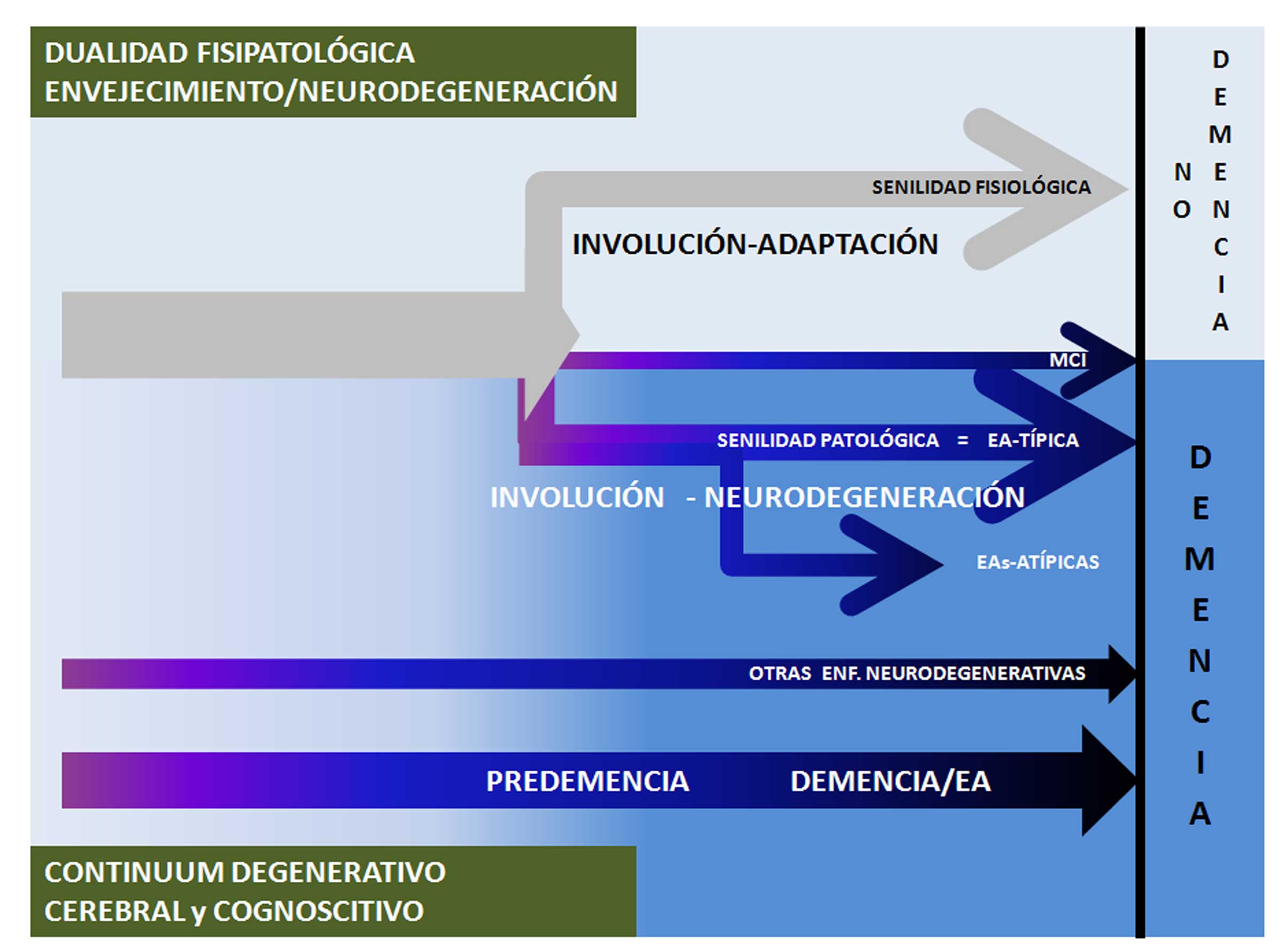
Figura 2. Senilidad fisiológica y senilidad patológica del cerebro. Existen dos distintas maneras de considerar el desarrollo de estos dos procesos de envejecimiento: 1) Por una parte se puede pensar que existe una involución senil que no conduce a la demencia y donde existen, además, signos de adaptación neuronal, glial y vascular que compensan muchos de los déficits morfofuncionales que se producen con la edad; mientras que por otra parte existe una involución que conduce a la demencia y donde predomina la neurodegeneración, lo que denominamos Enfermedad de Alzheimer típica (EA). Al lado de este proceso degenerativo se sitúan otros procesos más o menos relacionadas (EAs atípicas), que también cursan con demencias graves, y otros procesos con alteraciones cognoscitivas leves pero que nunca llegarán a recibir el diagnóstico de demencia (MCI; alteraciones cognoscitivas leves). 2) Por el contrario, se puede considerar que todos los individuos sufren un proceso de degeneración cerebral continuado (“continuum”) en el que se transita desde un estadio involutivo senil benigno, sin demencia, hasta la demencia tipo EA más grave pasando por etapas de predemencia con alteraciones morfofuncionales cada vez más graves.
3. ¿Existe un continuum entre la involución senil normal o fisiológica y la involución senil patológica tipo Alzheimer?
En la introducción se apuntaba la gran importancia práctica de conocer si existe una continuidad entre la senilidad normal y la patológica en base a que la demencia típica de la segunda plantea graves problemas sanitarios y sociales. Pero también, teórica y prácticamente, sería de enorme importancia para entender la patogenia y desarrollar terapéuticas para las diferentes involuciones seniles del cerebro. Conocer los mecanismos que condicionan un paso tan grande de la normalidad a la anormalidad senil en caso de que existiese, sería de una gran ayuda para atender a los humanos en su proceso de envejecimiento. Una de las cuestiones más polémicas en este tema es la existencia o no de síntomas o signos “patognomónicos” de EA, es decir que sean indubitadamente indicativos de que un enfermo padezca Alzheimer (o había padecido, en el caso de estudios post-mortem de los cerebros) porque no aparecen en el caso de los ancianos “normales”.
Se ha dado el nombre de continuum al teórico desarrollo evolutivo (involutivo) que lleva desde una “normalidad” morfofuncional del cerebro a una “anormalidad” (“o situación patológica”) del mismo (37-40) (Figura 2). Es decir, el continuum fisiopatológico equivale en el plano clínico al avance evolutivo del deterioro cognoscitivo que conduce desde la normalidad a la demencia (Figura 2). Por supuesto, los inicios de este continuum y las posibles etapas (fases o gravedad de la demencia) por las que discurre el avance del deterioro son tan imprecisos como los términos de “normalidad”, “anormalidad”, “deterioro o alteración leve, moderado, grave, severo”, etc., que se emplean en lenguaje médico y quedan en la mayoría de los casos a criterio del juicio clínico-diagnóstico del profesional a cargo del enfermo. En el mundo anglosajón, bien representado por el DMS III/IV/V, se intenta llegar a diagnósticos más objetivos a través del análisis estadístico de los resultados de las pruebas, especialmente las neuropsicológicas (cognoscitivo-comportamentales), que nos señalan los “puntos de corte” para separar los individuos “normales de los anormales” o los situados en una fase u otra más avanzada de la demencia (Figura 3). Estos puntos de corte se definen por consenso y, en realidad, no establecen con exactitud un diagnóstico sino que indican una probabilidad del mismo que el futuro seguimiento del enfermo, o su estudio post-mortem, se encargará de confirmar.
Lo que sí avala el estudio clínico del deterioro cognoscitivo de las poblaciones, con muestras suficientemente grandes y representativas, es que, con la aplicación de cualquier prueba neuropsicológica mensurable, la representación gráfica corresponderá a una curva de progresión o regresión continuada, lo que parece confirmar el continuum en el deterioro cognoscitivo (Figura 3).
La confirmación morfofuncional (neuropatológica, histoquímica, bioquímica celular y molecular, genética) de la existencia del continuum es un tema de intenso debate aunque poco estudiado en sí mismo. Aunque algunos aspectos se tratarán más adelante conviene ya señalar las grandes discrepancias de opinión al respecto basados tanto en los resultados obtenidos con distintas técnicas como en la interpretación de resultados empleando las mismas técnicas, además de la absoluta imprecisión morfofuncional de definir cualquier fase o estadio que teóricamente se presupone que existe desde la “normalidad” absoluta hasta la patología de la EA terminal.
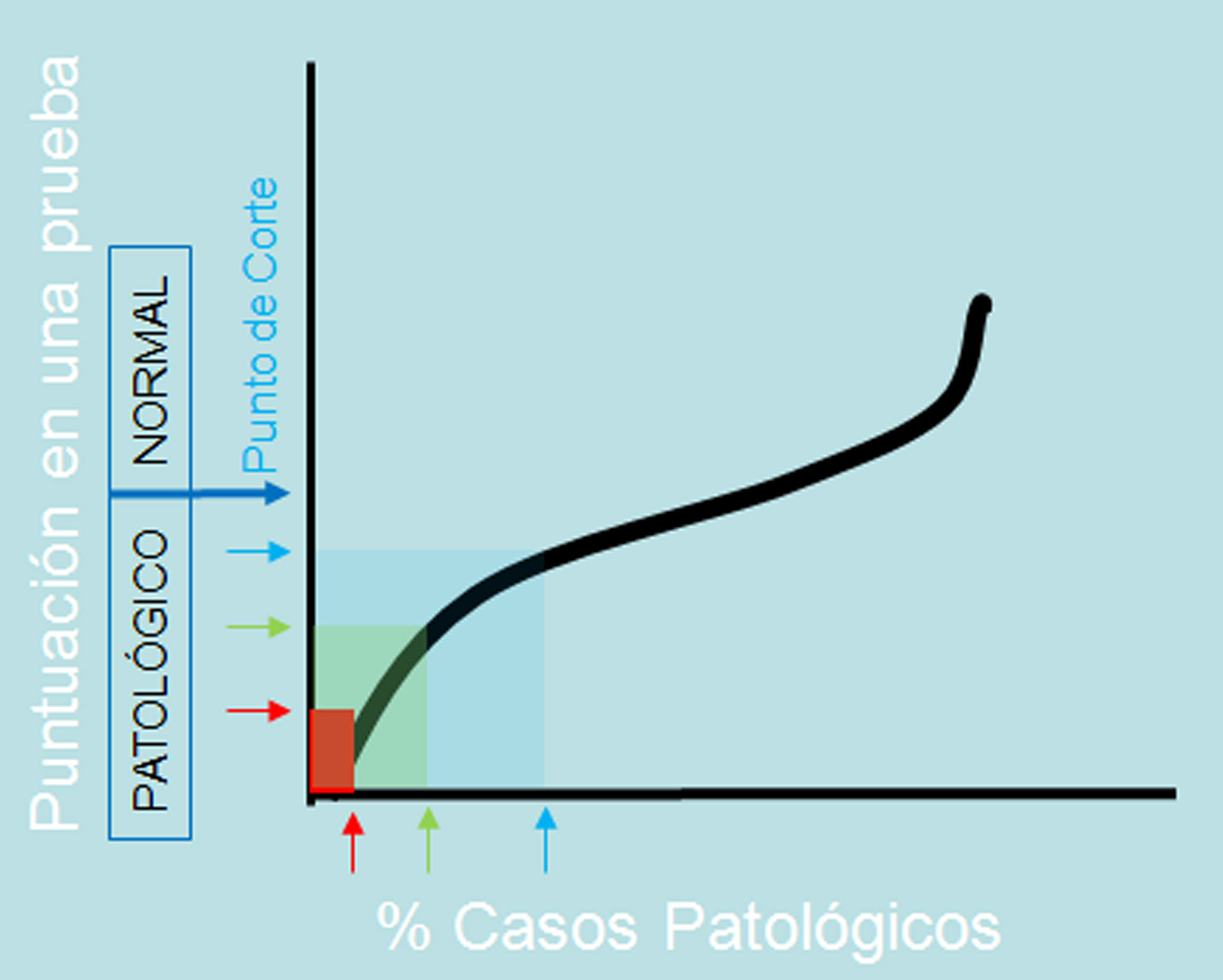
Figura 3.- Los atributos de “normal” y “anormal/patológico” que se asignan a los individuos o a los cerebros post-mortem que se estudian varían según los criterios que se apliquen. El “punto de corte” entre los dos posibles atributos normal/patológico que se puede aplicar a una población en un estudio determinará el porcentaje de casos con uno u otro diagnóstico. En el esquema se ejemplifica esto en una gráfica donde se representa la puntuación de una población en una prueba neuropsicológica teórica donde se pretende determinar el grado de demencia (normal o patológico) en base al número de aciertos en las pruebas que se le realizan. Dependiendo de que el “punto de corte” (puntuación, que se ha acordado por consenso, para diferenciar los individuos normales de los patológicos) sea más o menos exigente, variará el porcentaje de casos diagnosticados como patológicos.
Tomemos como ejemplo los dos principales tipos de lesiones “características” de EA conocidas desde la época de Alzheimer, los ovillos neurofibrilares (acumulaciones intraneuronales de proteína Tau, especialmente altamente fosforilada) y los depósitos amiloideos extracelulares de diferente morfología y composición. En primer lugar, al no ser estas lesiones “específicas” de la EA (ya se dijo que no son patognomónicas) el diagnóstico EA no puede establecerse por la sencilla demostración de su existencia por cualquier método, sino por la densidad de estas lesiones en regiones preestablecidas del cerebro. En segundo lugar, se ha demostrado que en muchos cerebros “normales” (los “controles” en los estudios sobre la EA), sin sintomatología de demencia, existían muchas alteraciones neuropatológicas señalados como características de EA y por tanto se ha recurrido a fijar criterios diagnósticos basados en el “número” y no en el “tipo de lesiones. Por ello ha sido necesario que se definieran protocolos de diagnóstico, en reuniones de consenso de expertos, para “asegurar” la mayor probabilidad de acierto diagnóstico. En estos protocolos se acuerda el tipo de técnica a utilizar en el diagnóstico (impregnaciones metálicas tipo Bielschowsky, plata-metenamina o Gallyas; reacciones inmunohistoquímicas contra proteína tau/tau-fosforilada y beta-amiloide; reacciones especiales con colorantes como tioflavina-S o Rojo Congo), el tipo de lesión a considerar o cuantificar (especialmente los distintos tipos de “neuritas distróficas” - prolongaciones axónicas o dendríticas neuronales anómalas - con depósitos alterados de proteína tau, así como las diferentes “placas” amiloideas con o sin “core” denso, con corona o no de neuritas distróficas y los acúmulos de amiloide difuso. Figura 4), el número de estas lesiones presentes en regiones seleccionadas del cerebro, y las regiones que deben seleccionarse para el estudio (corteza prefrontal, parietal y temporal; hipocampo; corteza cingulada; etc.) (30, 31, 41). A mayor abundamiento, los protocolos diagnósticos incluyen también como factor importante de corrección a la edad, lo que supone una aceptación de que con el avance de la edad aparecen lesiones patológicas aunque no sean indicativos de demencia o EA. Pues bien, a pesar de todos los avances de la investigación, todavía no se ha definido con exactitud cuáles son las cascadas de acontecimientos neuropatológicos en el transcurso del deterioro cognoscitivo clínicamente detectable ni que acontecimientos podrían marcar una fase determinada de este deterioro que se corresponda con la clínica (10-12, 42, 43). Es decir, la progresión del deterioro cognoscitivo a través de un continuum no ha podido ser confirmada o negada por los estudios neuropatológicas ya que no existe una clara y total correlación clínico-patológica del avance de la enfermedad. Los más prometedores estudios llevados a cabo por Braak y Braak (44) mostraron que existía una secuencia congruente de progresión neuropatológica de la EA con las características del deterioro cognoscitivo: la aparición de neuronas con ovillos neurofibrilares (Tau/Tau-fosforilado inmunopositivos) en el cerebro seguía una secuencia definida de seis fases de progresión espacial (grados I a VI de Braak y Braak) (Figura 5) conforme avanzada el grado de demencia, aunque no se pudiera predecir in vivo el grado Braak y Braak por estudios neuropsicológicos. Por el contrario, a pesar de que la teoría amiloidea de la EA ha sido la que goza y ha gozado de mayores defensores y seguidores, nunca se ha podido demostrar una correlación clínico-patológica entre el grado de demencia y la existencia de tipo y/o número o localización de lesiones amiloideas (45, 46). Así mismo, cada vez está mejor documentado como cerebros de individuos “control”, “sanos” o “no dementes”, presentan características “anormales” en el estudio de muchos parámetros morfofuncionales (47, 48).
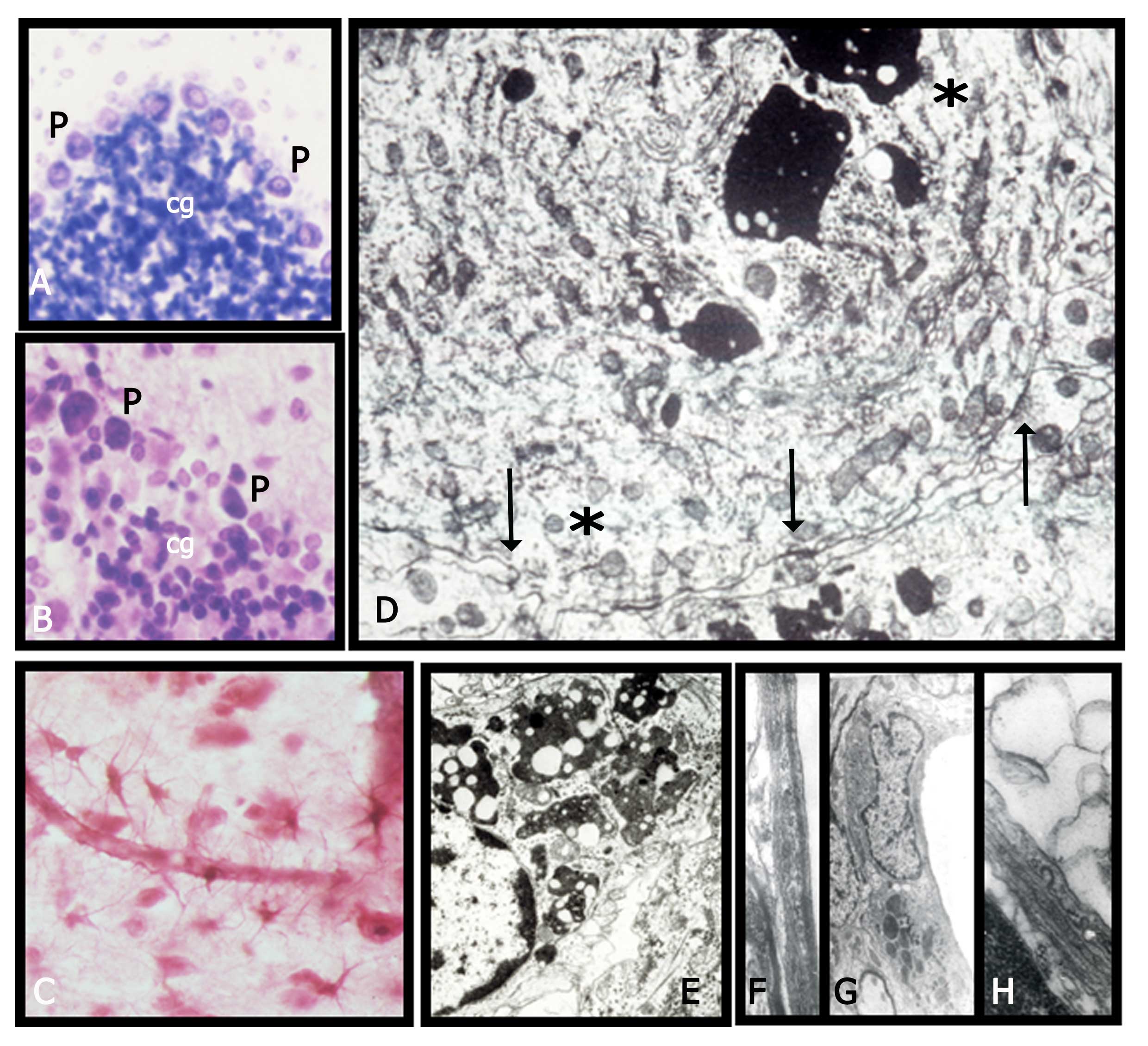
Figura 4a.- Alteraciones morfofuncionales seniles comunes en la involución cerebral senil fisiológica y patológica. De manera general puede decirse que existe pérdida neuronal y distrofia (o signos genéricos de envejecimiento celular) de las células remanentes. En A y B se muestra una laminilla cerebelosa adulta y senil, respectivamente, donde se aprecia la pérdida de neuronas de Purkinje (P) y de los granos (cg), así como las alteraciones distróficas de las células supervivientes, especialmente en las células de Purkinje que, al Microscopio Electrónico, evidencian (D) acumulación de lipofuscina, zonas de pérdida de organoides como Retículo Endoplásmico Rugoso y mitocondrias (*) y disminución de sinapsis (→). También sufren cambios, generalmente hipertróficos o distróficos (gliosis) las células astrogliales (C, con aumento de prolongaciones) y microgliales (E, con aumento de lipofuscina). Los vasos sanguíneos (en F, se observa la pared de un vaso normal), también sufren involución y muestran alteraciones como engrosamiento de la membrana basal (H) o signos de acumulación de substancias de desecho (G, acumulaciones lipídicas).
Los criterios diagnósticos han ido variando a lo largo de los años en parte por la aparición de técnicas más específicas para la demostración de proteínas amiloideas y tau y en parte, lo que es más importante en esta revisión, porque han ido variando los conceptos sobre la EA (5, 6). Todo ello nos sitúa también en la indefinición del inicio del presumible continuum fisiopatológico senilidad normal - EA. Otros tipos de cambios morfofuncionales que pueden observarse en cerebros normales o patológicos se tratarán a continuación.
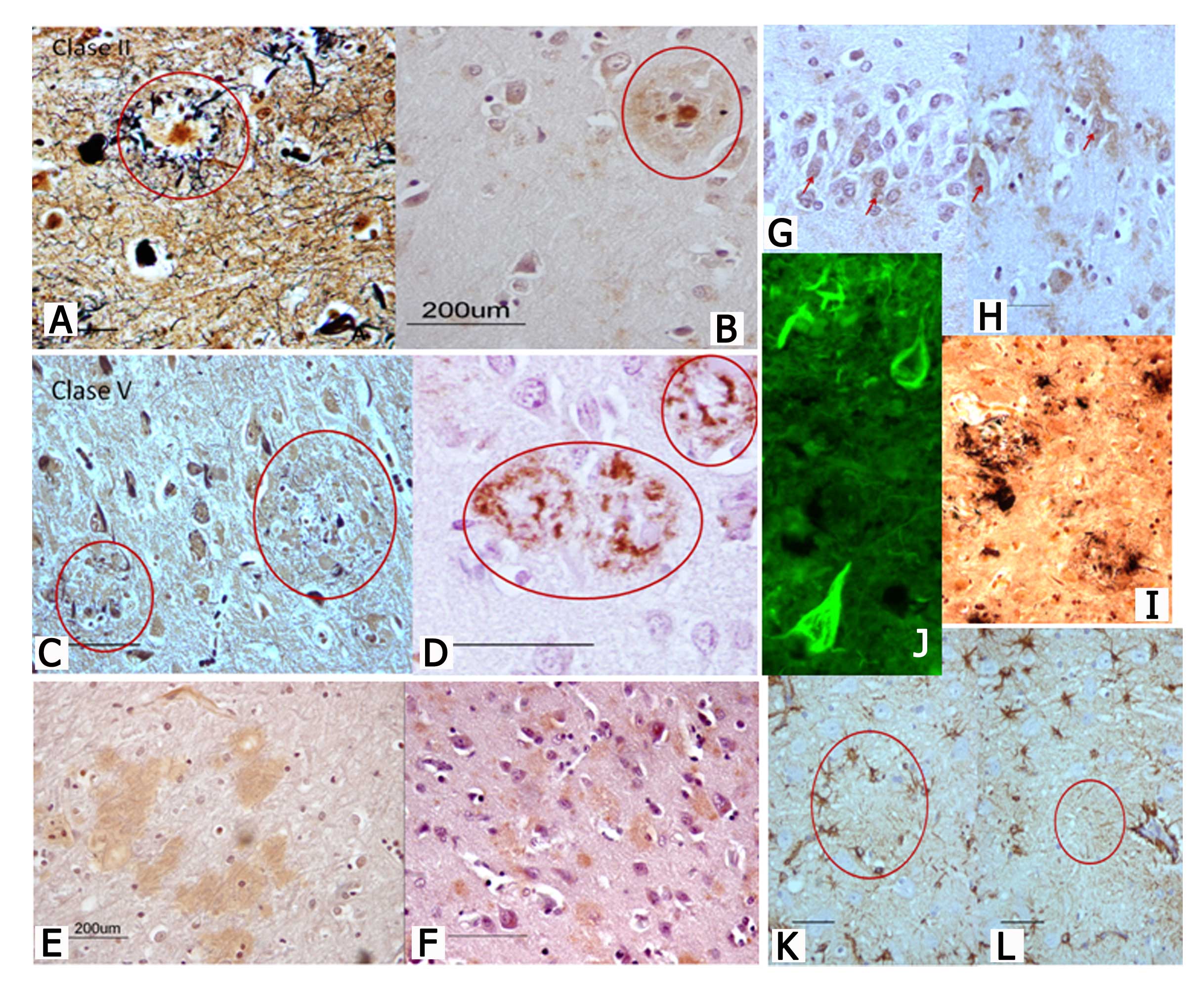
Figura 4b. Alteraciones neuropatológicas típicas del envejecimiento patológico / Enfermedad Alzheimer. A-F. Depósitos amiloides en “placa”. A, C y E, impregnaciones metálicas (Bielschowsky), y B, D y F, inmunotinciones con anticuerpos antiamiloideos. Depósitos estructurados de diferente tipo en corteza frontal: en A y B, placas con “core” y corona amiloide que con la impregnación metálica permite visualizar una gran cantidad de “neuritas” distróficas que no se observan con la inmunotinción por no contener amiloide, pero sí depósitos de proteína tau aberrante. En C y D, placas multilobuladas con condensaciones de amiloide y otras masas no bien tipificadas. En E y F, “placas difusas” de amiloide de contenido granular y límites poco precisos (“masas algodonosas”). En G(gyrusdentatus) y H (corteza prefrontal) se muestran otros tipos de depósitos amiloideos no estructurados, o “difusos”, en el neuropilo, así como depósitos intraneuronales. Todos estos tipos de depósitos diferentes parecen señalar la existencia de diferentes vías de generación de neuropatología amiloidea en el hombre durante el envejecimiento patológico, aunque la demencia EA se la vía final común (43). En J, se muestran los ovillos neurofibrilares en dos neuronas y “neuritas” distróficas en una placa amiloidea. En I, se observan células de microglía (teñidas mediante inmunorreacción frente al antígeno CD45) cuyas prolongaciones discurren por el neuropilo dela corteza parietal y penetran en las placas amiloideas. En K y L, se observan astrocitos hiperreactivos contra proteína glialacidófila (signo de astrogliosis) en neuropilo de la corteza prefrontal. Su relación con las “placas” amiloideas (marcadas con círculos) es variable, observándose una “corona” glial en K que no existe en la placa de la imagen L.
El término de continuum ha sido muy utilizado en el entorno de la EA para referirse a patogenia, evolución clínica, atención al enfermo, etc., (49, 50) ya que esta enfermedad o síndrome se caracteriza por la “progresión” de su curso evolutivo-involutivo que se manifiesta tanto porque a los síntomas iniciales en algunas áreas cognoscitivas se han agregado déficits de otras áreas hasta una completa involución de las funciones cognoscitivo-comportamentales, como porque los déficits de todas las áreas progresan lentamente aunque de manera independiente. La CIE-10 considera que el diagnóstico de EA se debe emitir sólo después de un período de seis meses en el que se compruebe que la supuesta EA ha progresado desde el grado de demencia cuantificado en el primer estudio clínico.
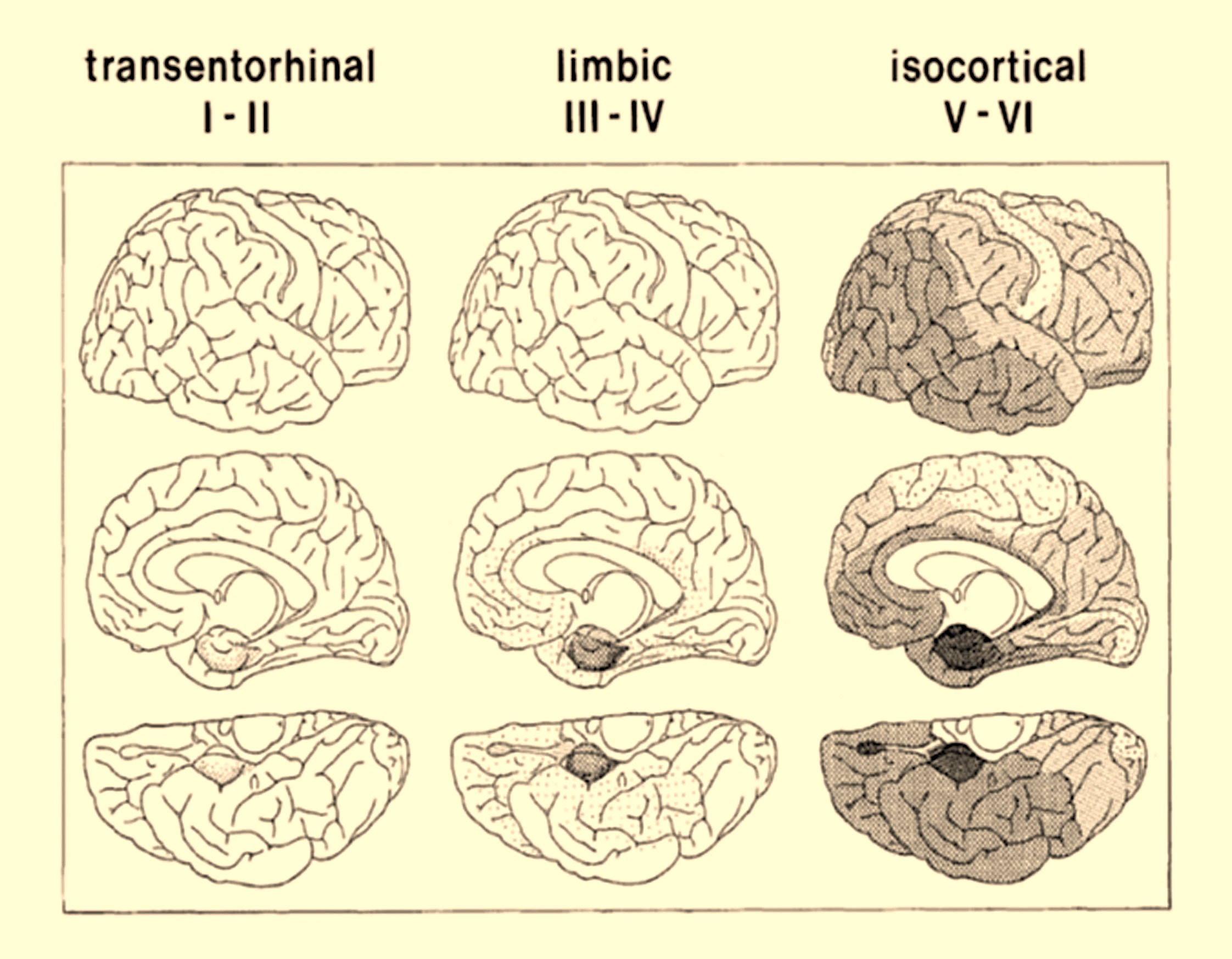
Figura 5.- Distribución del patrón de los cambios neurofibrilares (ovillos neurofibrilares y neuritas en neuropilo). Se diferencian seis estadios. En los estadios I y II las lesiones están prácticamente confinadas a la región transentorrinal. Los estadios III y IV, también llamados límbicos, presentan importantes alteraciones en la corteza límbica. Los estadios V y VI muestran alteraciones “isocorticales”. (Modificado de ref. 44).
4. Similitudes y diferencias entre la involución senil cerebral normal y patológica
Si la existencia o no de alteraciones neuropatológicas (depósitos de amiloide, depósitos de proteína Tau anómala en somas -ovillos neurofibrilares- o dendritas distróficas -neuritas) es un importante (aunque impreciso) hecho para concluir si un cerebro es “normal” o “anormal”, los cambios morfofuncionales que se presentan conforme avanza la edad son fundamentales tanto para entender la fisiopatología del envejecimiento como para analizar si existe una posible continuidad o secuencia de presentación y evolución de alteraciones entre el cerebro senil normal y el patológico.
CUADRO II
PRINCIPALES SIMILITUDES Y DIFERENCIAS DE ALTERACIONES ENTRE SENILIDAD NORMAL Y PATOLÓGICA (50-70, 90-118) SIMILITUDES - DISTROFIA Y ATROFIA NEURONALES - PÉRDIDA NEURONAL, APOPTOSIS - LIPOFUSCINOSIS Y ALTERACIONES DE ORGANOIDES NEURONALES - DISMINUCIÓN DE SINAPSIS Y NEUROTRANSMISIÓN - FENÓMENOS (NEURO)INFLAMATORIOS Y ESTRÉS OXIDATIVO - DISMINUCIÓN DE SISTEMAS DEFENSIVOS - DISMINUCIÓN DE SISTEMAS ADAPTATIVOS - INVOLUCIÓN DE SISTEMAS DE ACTIVACIÓN/REGULACIÓN DE LOS NÚCLEOS BASALOCORTICALES Y RETICULARES TRONCOENCEFÁLICOS (COLINÉRGICOS, ADRENÉRGICOS, DOPAMINÉRGICOS, SEROTONINÉRGICOS) - INVOLUCIÓN DE SISTEMAS TRÓFICOS (NGF, BDNF) - AUMENTO DE SISTEMAS DE INVOLUCIÓN Y MUERTE NEURONAL (CITOQUINAS, CASPASAS, PROTEÍNAS PRO-APOTÓTICAS) - ALTERACIONES DE CELULAS GLIALES y VASOS SANGUÍNEOS DIFERENCIAS - ACUMULACIONES PROTEICAS (DEPÓSITOS DE AMILOIDE - PLACAS, AMILOIDE DIFUSO Y AMILOIDE PERIVASCULAR-, DEPÓSITOS DE PROTEÍNA TAU -OVILLOS NEUROFIBRILARES Y NEURITAS DISTRÓFICAS-, ACUMULACIONES DE SINUCLEÍNA, UBIQUITINA, etc) - PÉRDIDAS COGNOSCITIVAS (DEMENCIA) y TRANSTORNOS COMPORTAMENTALES - PRESENCIA DE ISOFORMAS PATOLÓGICAS DE GENES RELACIONADOS CON LA ENFERMEDAD DE ALZHEIMER (dominantes: APP, Presenilinas 1 y 2; de susceptibilidad: APOE-4, CR1, ABCA-7, CD2AP, …, más de 200) |
En el Cuadro II se muestran algunos de los cambios más llamativos que se han descrito en humanos seniles con o sin demencia. Frente a una larga lista de similitudes se marcan una pequeña relación de diferencias: acumulaciones proteicas amiloideas, ovillos neurofibrilares y neuritas distróficas Tau-inmunopositivas, depósitos de alfa-sinucleina; isoformas especiales de algunos genes de susceptibilidad. Sin embargo, estas diferencias, como ya se ha dicho, no son per se patognomónicas de senilidad patológica/EA ya que ninguna es específica de un tipo de cerebro patológico: en bastantes cerebros seniles “normales” desde el punto de vista del deterioro cognoscitivo, aparecen depósitos de amiloide y/o Tau (51, 52) y, por otra parte, en demencias no EA existen también alteraciones que se consideraban hasta hace poco características de la EA, como los depósitos relacionados con la proteína Tau existentes en todas las tauopatías o taupatías (35, 36). Únicamente en la EA familiar existen genes específicos cuyas isoformas condicionan la aparición de la enfermedad demencial (Cuadro II) pues en la EA esporádica, muchos genes de susceptibilidad (APOE,) presentan isoformas que propician la aparición de demencia pero sin que se pueda precisar la fecha de comienzo, curso o intensidad de la patología (53, 54). Pero se puede decir que el cuadro neuropatológico del cerebro senil patológico es similar al descrito en la mayoría de los libros sobre la EA y en las introducciones de los trabajos de investigación sobre esta enfermedad, donde se menciona la alta densidad de depósitos de amiloide y tau, la involución del sistema colinérgico basalocortical, la astrogliosis y microgliosis, el aumento del estrés oxidativo y de los elementos celulares y moleculares del proceso neroinflamatorio, la pérdida o disfunción de las sinapsis y la involución de los sistemas neurotróficos/adaptativos frente al incremento de los sistemas de disfunción/muerte celular (25, 41, 44, 55-60).
Todas las alteraciones comunes a los dos procesos de envejecimiento (y a las entidades de envejecimiento en el límite demencia/no demencia y de EAs atípicas), existen en mayor o menor intensidad en cada proceso (aunque con mayor gravedad en la EA), pero no puede establecerse un nivel o tipo característico de involución/adaptación senil fisiológica o de neurodegeneración patológica en ninguno de ellos de una manera objetiva ni con certeza absoluta. Entre estas alteraciones comunes se encuentran: pérdida neuronal más o menos acusada en distintas regiones corticales, subcorticales y troncoencefálicas, disfunciones de sistemas basalocorticales, signos de apoptosis neuronal y elevación de sus marcadores- caspasas, proteínas pro-apoptóticas-, lisis de organoides subcelulares, acumulación de lipofuscina, alteraciones de la glía (astrogliosis y microgliosis) y de los vasos (acumulaciones de substancias en lisosomas secundarios, engrosamiento de endotelio y membrana basal, esclerosis y trombosis vascular) (61-64). Hay que señalar que no todas las alteraciones se presentan en todos los centros nerviosos y que hay regiones, como el cerebelo y la corteza occipital, que parecen ser las menos vulnerables, muestran pocas diferencias involutivas, mientras otras ponen de manifiesto grandes diferencias de involución senil entre los cerebros seniles normales y patológicos, precisamente en las regiones más castigadas en la EA (hipocampo, cortex prefrontal, estructuras límbicas, núcleos basalocorticales) (25, 30, 31, 65). Aunque de una manera más teórica que científicamente comprobada, da la impresión de que sobre un substrato de cambios morfofuncionales moleculares y celulares se van desarrollando cambios patológicos conforme disminuyen los mecanismos de defensa del organismo y del propio CNS así como disminuyen los mecanismos de adaptación de las neuronas ante los insultos externos e internos (Figura 6) (66).
Las conexiones sinápticas disminuyen en gran número de circuitos, sin embargo, la pérdida de conexiones no se corresponde con la pérdida o disfunción de neuronas: en el envejecimiento senil fisiológico existe una importante neosinaptogénesis que suple la pérdida de neuronas (Figura 1) (24, 66). Como ejemplo puede citarse la proliferación de terminales axónicos en hipocampo a partir de fibras remanentes y la neoformación de espinas dendríticas en las neuronas supervivientes para reforzar la conectividad (fenómeno similar al logrado experimentalmente en el aprendizaje y con entrenamientos tanto en animales jóvenes como seniles - 23, 24) (Figura 1).
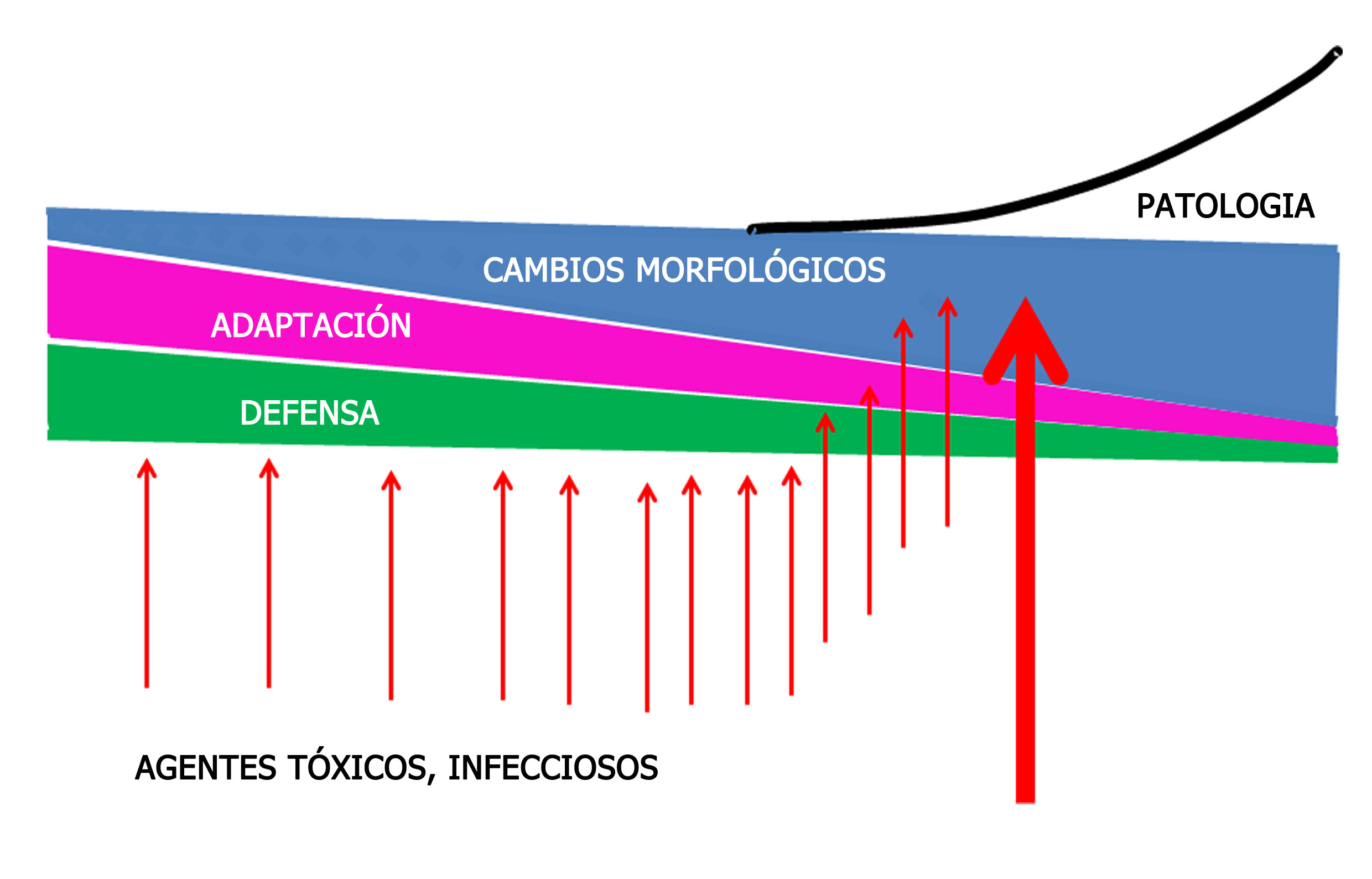
Figura 6.- Esquema que intenta resumir los posibles cambios generales acaecidos en el cerebro para propiciar el desarrollo de patología durante la senilidad. El cerebro (sus neuronas y sus células gliales) poseen sistemas y mecanismos de defensa frente a las agresiones externas e internas que evitan cambios morfofuncionales en el mismo y mecanismos de adaptación para prevenir la involución senil. Conforme avanza la edad disminuye la defensa y la adaptación con lo que se producen cambios morfológicos en neuronas y circuitos neuronales que son la manifestación de la degeneración senil. Si los agentes tóxicos en infecciosos, externos e internos rompen las barreras de defensa y adaptación, se desarrolla una involución patológica
Los sistemas neurotransmisores también presentan alteraciones seniles, fisiológicas y patológicas. En general existe una menor involución en la síntesis de receptores que de neurotransmisores, lo que es de utilidad en los posibles tratamientos con agonistas, siendo especialmente vulnerables los de las neuronas reticulares basalocorticales (colinérgicos, adrenérgicos, dopaminérgicos y serotoninérgicos) (67) (Figura 7). La involución colinérgica se ha invocado tanto para fundamentar la “disfunción geriátrica” (especialmente centrada en alteraciones de la memoria que no se corresponden con un cuadro de demencia - 61) como para explicar la EA. La teoría colinérgica de la EA es la base del tratamiento anticolinesterásico de la enfermedad (61, 68, 69), que junto a la memantina, bloqueante de receptores NMDA para glutamato, son los únicos tratamientos aprobados en la actualidad contra la EA (70). La disfunción colinérgica puede basarse en uno o varios de los siguientes problemas: a) pérdida o disfunción de las neuronas de los núcleos colinérgicos basalo corticales (septum, banda diagonal de Broca, nucleus basalis mognocellularis o núcleo de Meynert de los primates, incluido el hombre – Figura 7, 8); b) disminución o disfunción de los axones terminales corticales que hacen disminuir la acetilcolina que mantiene el nivel óptimo de excitación de las neuronas corticales; c) disminución del sistema neurotrófico específico NGF (“nerve growth factor”) que mantiene el funcionamiento y hace responder correctamente al sistema colinérgico basalocortical (Figuras 7, 8). Esta involución colinérgica parece manifestarse con una intensidad leve a moderada en el envejecimiento fisiológico y con una intensidad moderada-grave en el envejecimiento patológico EA. En el Cuadro III y en la Figura 9, se muestran las variaciones de distintos parámetros morfológicos bioquímicos e histoquímicos del sistema colinérgico basalocortical en varias situaciones (desde el envejecimiento fisiológico a la EA terminal).
Respecto a la posible paradoja existente entre suponer que en el envejecimiento (fisiológico o patológico/EA) ocurre una bajada de neurotransmisión glutamatérgica, y emplear como tratamiento de la EA el bloqueo de receptores tipo NMDA para glutamato con el fin de limitar la funcionalidad de una parte de este complejo sistema de neurotransmisión, hay que decir que todavía quedan muchas cuestiones que aclarar sobre la posible implicación de la neurotoxicidad del glutamato, absoluta o relativa en ciertas regiones del SNC, en el origen y la progresión del envejecimiento y la EA así como de los beneficios y del mecanismo de acción del medicamento memantina, aunque haya sido internacionalmente aprobado para su uso clínico. Podría ocurrir que el bloqueo de los receptores NMDA, aunque empeorando la neurotransmisión glutamatérgica en ciertos circuitos, mejorara la concentración intracelular de calcio, que puede llegar a ser neurotóxica, al impedir el paso del catión a su través cuando se activan por el glutamato.
Para la mayoría de las investigadores, lo que caracterizaría al envejecimiento patológico (EA) sería la especial intensidad de las alteraciones junto a determinadas combinaciones de alteraciones, pero sólo en regiones específicas y tipos concretos de neuronas. Especialmente significativo es el caso de la corteza entorrinal donde parece que se producen los primeros cambios en la EA (etapa I de Baak y Braak – 44. Figura 5). Aparecen aquí los primeros ovillos neurofibrilares en las neuronas piramidales, y se detectan disminuciones del número de neuronas y del espesor de la corteza con diferentes técnicas in vivo e in vitro (47, 48, 71, 72). Ciertos estudios funcionales parecen indicar que disminuye también la conectividad sináptica y el flujo de la información a este nivel (72).
Uno de los últimos y más importante estudios para probar la hipótesis del continuum senilidad fisiológica - enfermedad de Alzheimer (EA) tanto clínica como neuropatológicamente se llevó a cabo en 2012 analizando 2.083 cerebros del National Alzheimer Coordinating Center obtenidos en autopsias desde 2005 a 2012 (55).
CUADRO III

Solo en 853 casos existió una buena correlación entre los datos demográficos y neuropatológicos y la valoración de la demencia mediante las pruebas del método Clinical Rating-Sum of Boxes (CDR-SB). Tanto las placas neuríticas como los ovillos neurofibrilares predijeron de modo independiente el grado de demencia obtenido por la aplicación del CDR. Pero también se asocian de forma independiente con el grado de deterioro cognoscitivo la enfermedad grave de pequeño vaso sanguíneo, severa la angiopatía amiloide grave y la esclerosis del hipocampo. Por el contrario, la educación recibida se mostró como un factor protector independiente fuerte contra deficiencias cognoscitivas. La causa de la demencia leve a moderada continúa siendo incierta en el 14% de los pacientes. Estos datos indican que las placas y los ovillos neurofibrilares contribuyen independientemente al deterioro cognoscitivo, pero que otras enfermedades, como las vasculares, simultáneamente modifican la patología y que, también puede modificarse la expresión clínica, p. e., con la educación recibida. Por lo tanto, múltiples etiologías concomitantes pueden inducir un daño cerebral con senilidad patológica pero siguen mostrando incertidumbre sobre la existencia del continuum basándose sólo en las correlaciones clínico-patológicas entre demencia y presencia de ovillos neurofibrilares y placas.
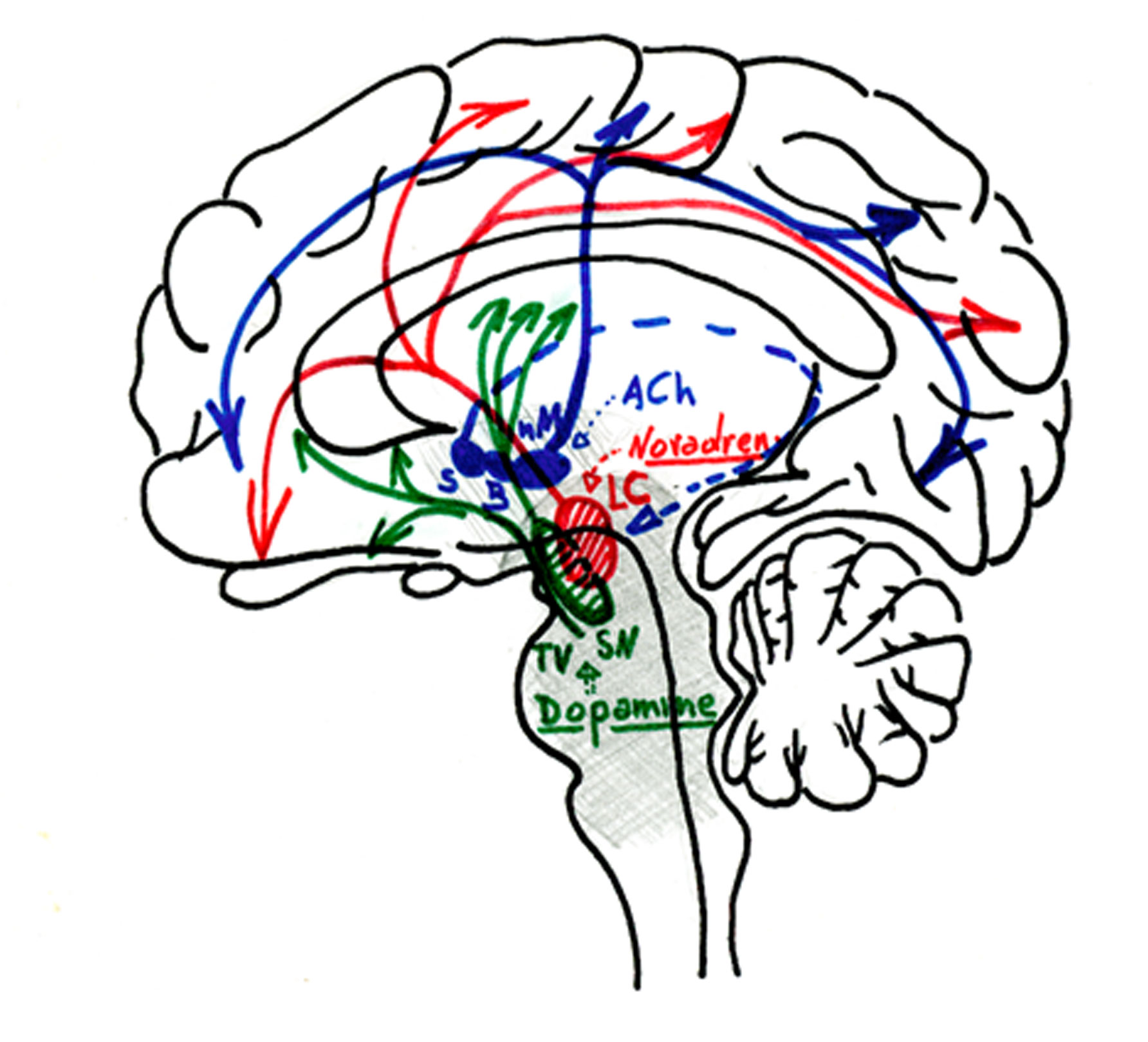
Figura 7.- Esquema donde se representan los sistemas basalocorticales y troncoencefálicos activadores que inervan la corteza cerebral del hombre (regiones sombreadas en gris): S= núcleos colinérgicos del septum que inervan, principalmente, el hipocampo; B= núcleos colinérgicos de la Banda Diagonal de Broca que inervan las áreas límbicas del cerebro; nM= núcleo de Meynert en los primates (equivalente al Nucleus Basalis Magnocellularis del resto de los mamíferos, donde residen los cuerpos de las neuronas colinérgicas que inervan el cortex cerebral); LC = locus coeruleus, donde se asientan las neuronas noradrenérgicas que inervan la corteza; SN= substancia negra y TV= substantia Tegmental Ventralis, donde se asientan las neuronas dopaminérgicas que inervan la corteza.
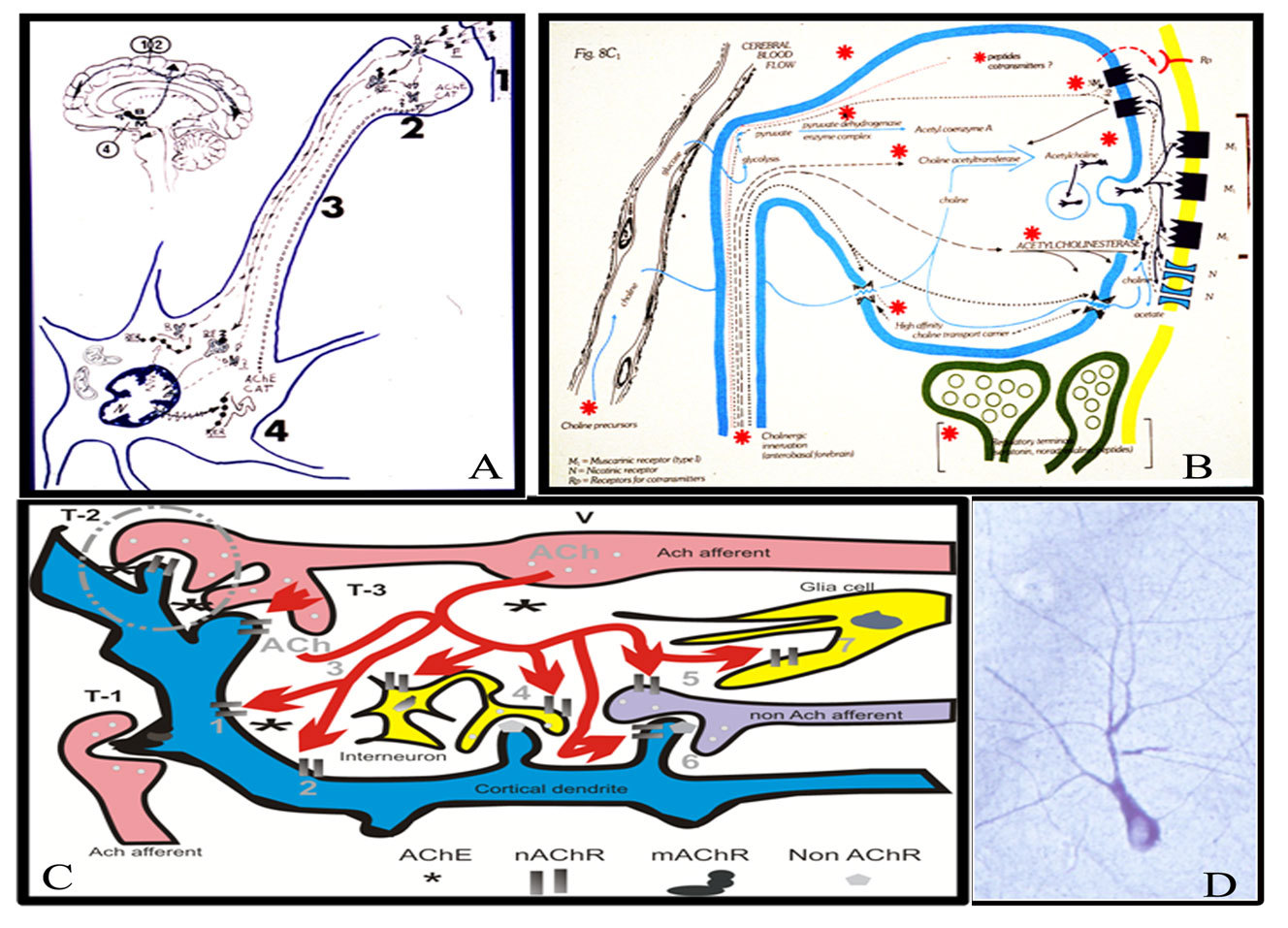
Figura 8.- Sistema Colinérgico basalocortical. Se recogen aquí diversos esquemas de los autores. En A, se representan los núcleos colinérgicos de la figura 7 (Septum, Banda Diagonal de Broca, Núcleo de Meynert) y una neurona colinérgica tipo con su soma en los núcleos basalocorticales (4) donde se sintetizan las enzimas colinérgicas CAT (colinacetiltransferasa), AChE (acetilcolinesterasa) que se transportan por el axón (3) hasta los terminales corticales (2) donde sintetiza y libera acetilcolina que actúa sobres las neuronas colinoceptivas (1) para mantener un estado de activación que facilite las respuestas óptimas. Estas neuronas corticales, así como las células astrogliales, sintetizan y liberan la neurotrofina NGF que es reconocida, captada y transportada retrógradamente por el axón (3) hasta el cuerpo neuronal, dónde actúa sobre el núcleo de la neurona para activar la síntesis de la mayoría de los enzimas y receptores del sistema colinérgico que se transportan a los terminales corticales. En B, se muestra con mayor detalle el sistema colinérgico cortical donde juegan un importante papel los receptores colinérgicos nicotinérgicos (N) y muscarínicos (M), tanto presinápticos como postsinápticos, que sirven tanto para activar las neuronas corticales como para regular la síntesis, liberación, acción, destrucción de acetilcolina y la recaptación de colina. La colina es captada desde los capilares o de los espacios interneuronales tras la destrucción de la acetilcolina por la acetilcolinesterasa y muchos terminales colinérgicos están regulados en su funcionamiento por otros terminales aminérgicos. En C, se amplía el esquema sobre la “inervación colinérgica basalocortical” mostrando la enorme complejidad de la misma. Las fibras colinérgicas (ACh; ACh aferente) liberan acetilcolina especialmente en varicosidades “no sinápticas” difundiendo entre las membranas neuronales (2 neurotransmisión de “volumen” o “no sináptica”, posiblemente un 80% de la acetilcolina sintetizada) y actúa sobre los receptores nicotinérgicos situados sobre dendritas y ramas de neuronas corticales en zonas no sinápticas (modificando las propiedades de estas neuronas), sobre interneuronas reguladoras y fibras aferentes no colinérgicas de la corteza y sobre células gliales. Otro porcentaje de acetilcolina se libera en terminales sinápticas que activan receptores colinérgicos nicotinérgicos y muscarínicos. El conjunto abarca mecanismos muy complejos y sofisticados que mantienen las neuronas corticales en óptimo estado de respuesta y producen en las mismas las modificaciones necesarias para llevar a cabo los complejos funciones que son necesarios en las funciones cognoscitivas. Estos sistemas degeneran durante la involución senil. En D, se observa una neurona colinérgica hipertrófica, superviviente y aislada, mantenida por sus mecanismos de adaptación en una región del núcleo de Meynert de un individuo con Alzheimer terminal (tinción, acetilcolinesterasa).
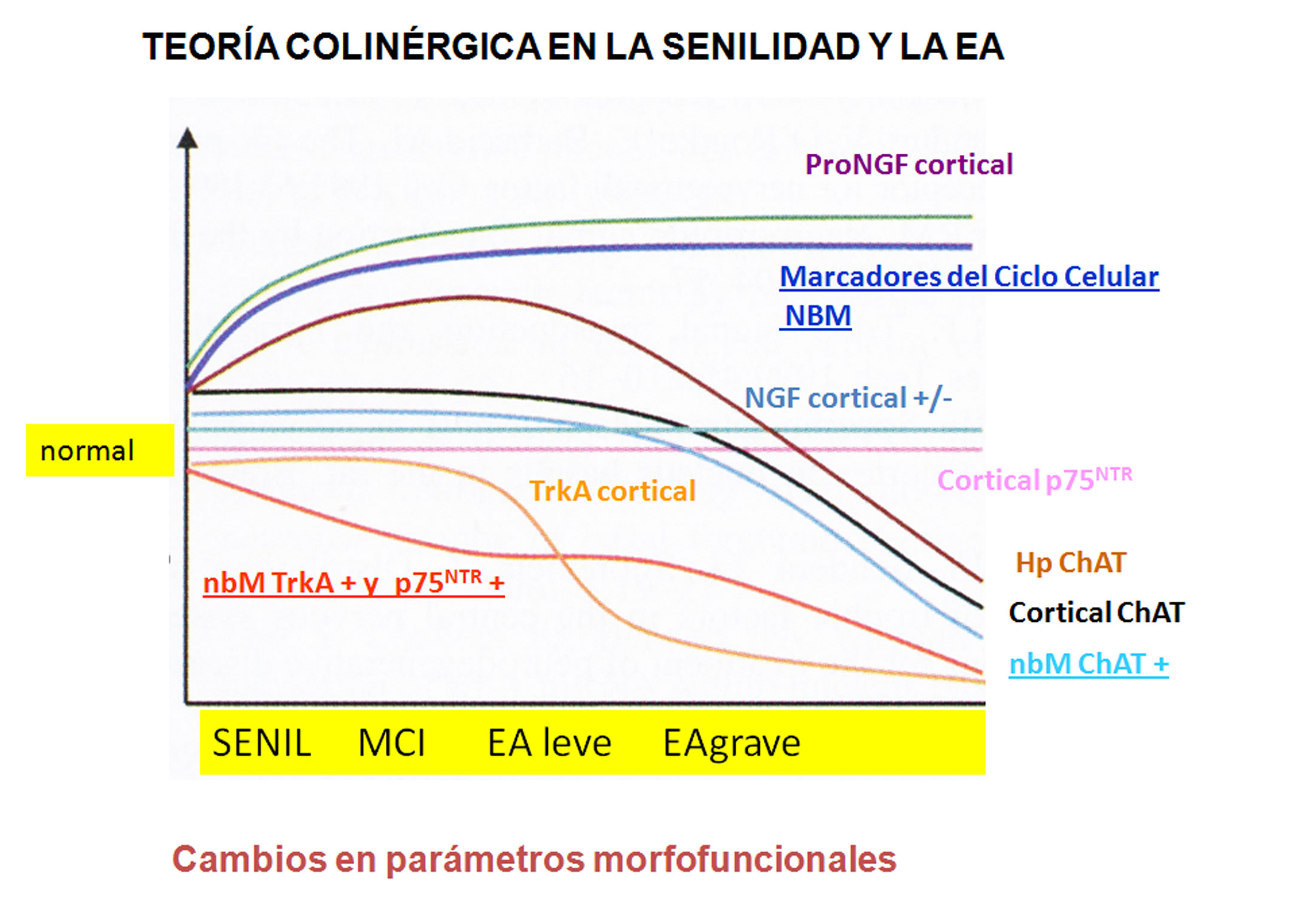
Figura 9.- Esquema sobre la evolución de los parámetros morfofuncionales en la teoría colinérgica de la senilidad y de la enfermedad de Alzheimer. Se representan algunas de los más importantes que relacionan la supervivencia y/o funcionalidad de la neuronas colinérgicas basalocorticales y del sistema NGF- neurotrófico que las mantiene con la posible evolución de la demencia Alzheimer desde la senilidad fisiológica (SENIL) al Alzheimer terminal (EA grave) pasando por el deterioro cognoscitivo leve (MCI) y el Alzheimer leve. A partir de la EA parece descender el número y/o función de las neuronas colinérgicas del núcleo de Meynert (nbM) aunque aumentar los marcadores del acto celular (indicio de degeneración neuronal) y la producción de pro NGF cortical (posible marcador de muerte celular) ya desde el inicio de la senilidad. La disfunción colinérgica se ofrecía en una disminución de síntesis de colinacetiltranferasa (ChAT) productora de acetilcolina y receptores para el NGF (TrkA y p 75NTR) en el nbM. En la senilidad fisiológica y MCI parece que los mecanismos de adaptación mantienen el nivel de ChAT en hipocampo (HpCAT). El NGF cortical y los receptores p75NTR para NGF corticales en la senilidad patológica/EA pueden o no sufrir disminuciones pero parece que el sistema colinérgico ya no mantiene funcionantes a las células. +/-= resultados de valormuy variable en diferentes estudios; += resultados muy repetitivos en la literatura científica. Comparar con el Cuadro III. (referencias 61, 66-70, 119, 120).
5. Estudios que pueden aclarar la existencia o no del continuum fisiopatológico en el envejecimiento cerebral
La respuesta definitiva a esta controversia vendrá dada por el conjunto de los resultados de estudios multidisciplinares en cerebros de diferentes edades y situaciones fisiológicas y patológicas, en especial: a) controles seniles normales, b) “centenarios “ humanos, c) casos que puedan ser considerados prototipos de patologías intermedias entre la senilidad normal y la patológica. Pero también será de gran ayuda estudios sobre el proceso de envejecimiento en: d) la evolución de los mamíferos (especialmente en los primates no humanos) y e) modelos experimentales donde se induce una patología Alzheimer, especialmente en ratones transgénicos con patología Alzheimer.
a) Controles seniles “normales”.
En todos los estudios sobre envejecimiento patológico/EA se incluye un grupo control “normal” de referencia. Este grupo control se “selecciona” de tal manera que se descarta cualquier síntoma o signo de posible patología por lo que muchas veces no representa el estado morfofuncional real del cerebro senil humano sino de grupos de individuos con especial resistencia a la involución senil. Además, el mínimo porcentaje de donaciones de cerebros o de voluntarios “sanos” en los estudios clínicos hace que sea bastante difícil llevar a cabo estudios con suficiente número de individuos para establecer teóricas fases del proceso de involución. Sin embargo, en los pocos casos en que se ha podido analizar un número significativo de individuos o de cerebros seniles “normales” sin demencia se ha apreciado un gran número de subconjuntos con muy diferente grado de alteraciones morfofuncionales que podrían ser totalmente calificadas de “anormales” o “patológicas” según determinados criterios (47, 48). En un trabajo de recopilación de datos sobre pacientes “control normal” del Berkley Aging Cohort (BAC) y del Alzheimer´s Disease Neuroimagen Iniciative (ADNI) (48), entre el 15 y el 20% de los casos tenían coeficientes “anormales” (EA) en parámetros como el espesor de la corteza cerebral, la captación de fluoro-deoxiglucosa (FDG-PET) y el volumen del hipocampo. Por el contrario, entre un 10 y un 25% de casos EA tenían cifras de “cerebro normal” en espesor de la corteza o consumo de glucosa, pero nunca presentaban valores normales de volumen hipocámpico (Figura 10). También estudios nuestros y de otros autores señalan porcentajes altos de “normalidad” neuropatológica en enfermos con demencia y de “anormalidad” en individuos no dementes (Figura 10). El estudio pormenorizado de estos individuos y/o cerebros post-mortem podían darnos las claves de cuáles son los cambios morfológicos y funcionales, y los mecanismos que los propician, en la transición del posible cerebro senil normal al patológico (continuum). Con ellas, podría pensarse en desarrollar estrategias preventivas contra la neurodegeneración desde la senilidad normal.
b) “Centenarios” humanos.
El “envejecimiento saludable” se aprecia en un elevado porcentaje de “centenarios” (personas de más de 90, 95 ó 100 años según diversos estudios) en diversas regiones del mundo. Aunque el porcentaje de estas personas mayores con funciones cerebrales muy conservados no es el mismo en todas las regiones, en todos los países existen bastantes casos, aunque parecen más llamativas las cifras de regiones con menores índices de supervivencia (islas del Caribe, Asia, Europa del Este). Algunos estudios, aunque sobre un número no significativo de casos, sobre estos individuos viejos (“old-old” o “very old”) y sus cerebros han encontrado alteraciones patológicas, aunque no muy intensas, en personas que no presentaban una clara sintomatología de demencia.
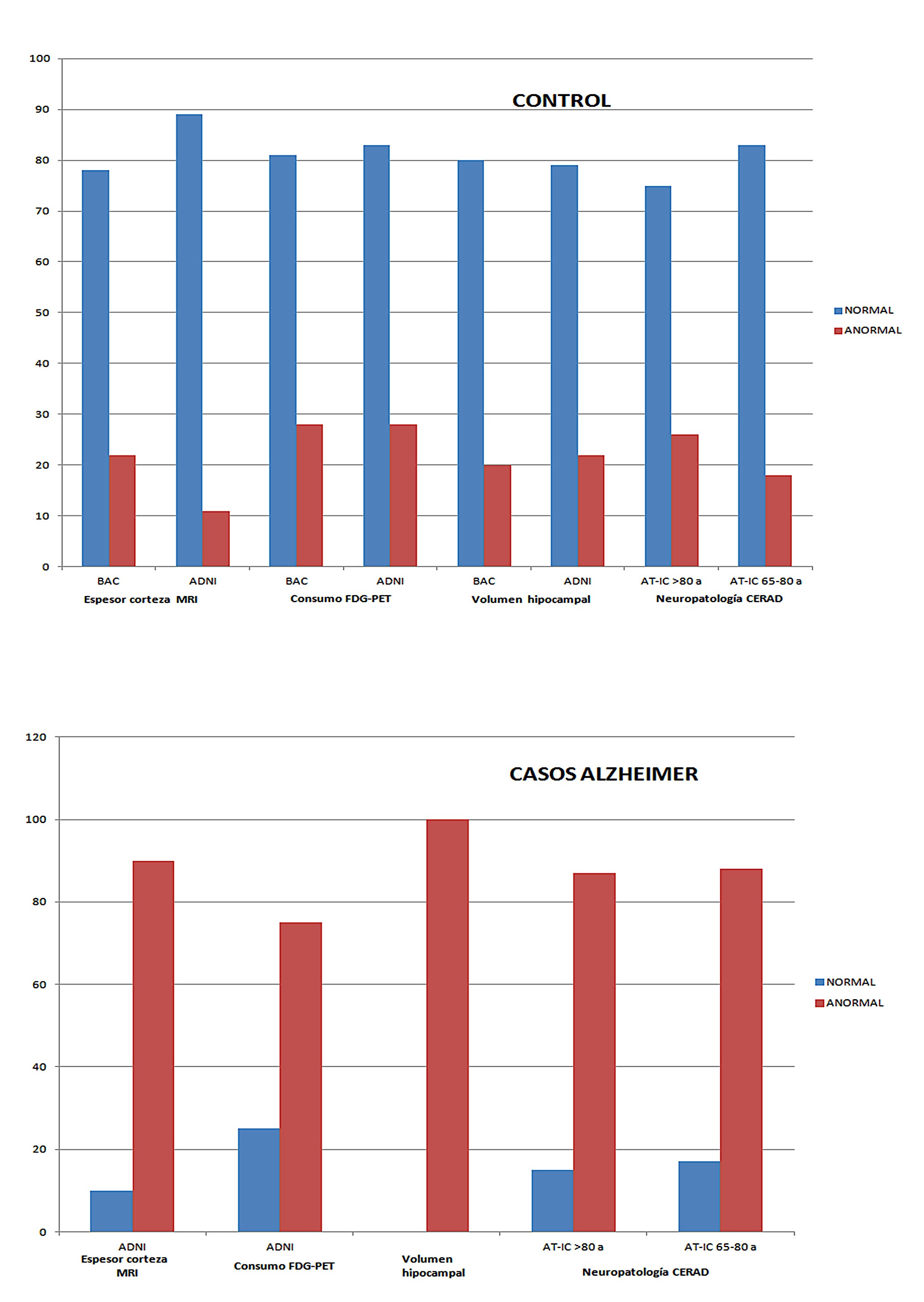
Figura 10.- Porcentajes de cerebros “normales” y “anormales” en diversos estudios, según los criterios actuales de consenso relativos a diversos parámetros tanto en grupos control senil normal como en grupos de senilidad patológica Alzheimer. Los grupos referenciados son los casos clínicos (ref. 48) de “BerkleyAgingCohort” (BAC) (72 casos) y “Alzheimer´s Disease Neuroimaging Initiative” (ADN) (286 casos) y los cerebros post-morten estudiados por los autores (155 casos). Los parámetros analizados son espesor de la corteza mediante resonancia magnética, consumo dedeoxiglucosa mediante tomografía de emisión de positrones (PET), volumen hipocampal mediante resonancia magnética y neuropatología Alzheimer según criterios de CERAD.
Esto parece confirmar que la presencia de alteraciones seniles que podríamos denominar patológicas no es indicativa de neurodegeneración o involución patológica/EA y avala también las resoluciones de consenso sobre la necesidad de que exista una clara densidad de alteraciones patológicas en el cerebro para un diagnóstico de “anormalidad” o patología EA. Sin embargo, nos deja sin precisar cuáles deben ser los límites exactos para separar la “normalidad” de la “anormalidad” porque no se han podido resolver los fundamentos de los cambios y de sus repercusiones. Así mismo, es de resaltar que en las personas mayores de 80 años que evolucionan hacia la EA (con signos de demencia), los cambios patológicos que se producen son menos graves que los de aquellos que inician la progresión hacia la EA en edades más tempranas, hacia los 65 años (73). En las pruebas neuropsicológicas se muestran menos “anormales” en las funciones ejecutivas mentales, en la atención y velocidad de procesamiento de la información y en la memoria inmediata. Así mismo, la disminución del espesor de la corteza en diversas regiones del cerebro (córtex cingulado, lóbulos parietal y temporal) no se aprecian diferencias frente a los ancianos “normales”. Todo esto hace que, a los niveles de discriminación diagnóstica actuales, sea muy difícil encontrar diferencias para la emisión de un diagnóstico de senilidad patológica/EA leve-inicial, pareciendo que existe una gran imbricación entre la situación morfofuncional de los cerebros “normales” y “patológicos” iniciales en los individuos mayores de 80 años.
c) Teóricos estadíos patológicos intermedios entre senilidad fisiológica y patológica/EA.
Los nuevos conceptos sobre la EA asumen la existencia de un envejecimiento patológico/EA que se inicia mucho antes de que aparezcan los primeros síntomas de demencia (EA prodrómica) (5, 6). El comienzo y las primeras fases de este proceso de envejecimiento es bastante difícil de precisar con las técnicas actuales ya que no disponemos de biomarcadores específicos de EA, ni centrales ni periféricos, ni tampoco la tecnología para detectar algunos de esos posibles biomarcadores. Las técnicas de neuroimagen más avanzadas (p.e., la deposición de amiloide mediante la técnica de la Tomografía de Emisión de Positrones –PET- que usa el compuesto de Pittsburg para unirse a estos depósitos -74, 75-, o la disminución del volumen o del espesor de la corteza o de determinadas regiones del SNC, especialmente el hipocampo y el lóbulo temporal, detectada con Resonancia Magnética nuclear -RNM) no pueden visualizarnos o cuantificarnos los primeros cambios cerebrales, pues precisan que se sobrepase el límite de discriminación. Por ahora, éste se sitúa siempre en fases intermedias-avanzadas del proceso de envejecimiento patológico, cuando ya la sintomatología de demencia ha propiciado un diagnóstico de EA leve/inicial. Por otra parte, se dispone de pocos cerebros de adultos “normales” de entre 30 y 60 años para su estudio y muy pocos de ellos se utilizan para detectar cambios sugerentes de degeneración o involución senil para que pueda concluirse de manera firme si existe un inicio hacia la senilidad patológica desde la madurez del estado adulto o bien desde la involución cerebral senil normal. En esta situación, la investigación de individuos o de cerebros que puedan ser definidos como pertenecientes a entidades patológicas intermedias entre la “normalidad” del adulto y la EA terminal, es de importancia capital. De momento sólo una entidad, la “alteración (o deterioro) cognoscitivo (o cognitivo) leve” (ACL o DCL) (“mild cognitive impairement”), parece estar ya asentada con criterios muy bien definidos para su diagnóstico y con definiciones bastante precisas de las alteraciones cognoscitivo/comportamentales y de algunos de los cambios en marcadores proteicos y de estrés oxidativo (7, 8). Esta entidad parece que un plazo de tres a cinco (o diez, según algunos otros autores) años evoluciona a EA, por lo que puede ser considerada como una fase intermedia de la involución patológica EA (8, 9).
Otros estadios intermedios en este curso patológico (o EA prodrómica o EA asintomática) todavía no han sido bien determinados, pero deben ser investigados aquellos subgrupos de personas que puedan aislarse de alguna manera clara cuando se hacen pruebas neuropsicológicas en los estudios epidemiológicos. En todas las pruebas neuropsicológicas se fijan “puntos de corte”, generalmente por consenso entre expertos para delimitar los individuos “normales” frente a los “anormales” de la población estudiada. Estos puntos de corte deberían ser revisados en profundidad para definir las subpoblaciones a estudiar y lograr avances en la patogenia del envejecimiento patológico.
d) Envejecimiento en la evolución de los mamíferos, especialmente de los primates no humanos.
Se ha dicho repetidamente que el problema más grave en el estudio de la EA es que no existe ningún mamífero que padezca EA de manera espontánea o inducida. Esta clásica aseveración ya no es totalmente cierta en la actualidad ya que existen algunos “modelos Alzheimer” en mamíferos que pueden ayudar a esclarecer el problema del envejecimiento normal y patológico/EA en el humano. Especialmente consideraremos en esta revisión dos modelos: uno natural, los primates no humanos, y otro experimental, los modelos transgénicos con neuropatología alzhéimer (tratados a continuación).
Aunque la proteína precursora de amiloide, APP, es una proteína ancestral de membrana que aparece evolutivamente en los primeros eucariotas y cuya función sigue sin estar clara (incluyendo su posible actuación como canal iónico, como proteína fijadora de metales como cobre, o de calcio, o como proteasa), no se presenta como fuente de patología hasta los mamíferos, especialmente de manera totalmente cierta en el hombre (76, 77). Además, a pesar de que su metabolismo por la vías “amiloidogénica” y “no amiloidogénica” (dependiente de la catabolización por la beta-secretasa o BACE- beta-site APP cleaving enzyme-, o por la alfa-secretasa) (78) existe ya en casi todas las especies de mamíferos, prácticamente no aparecen depósitos amiloideos en cerebros seniles más que en los primates. Son excepcionales los casos recogidos en la literatura de animales con amiloidosis cerebrales (oso polar, conejo), sin embargo muchas especies de primates de diferente grado de evolución (prosimios, simios del nuevo mundo y simios del viejo mundo) han mostrado la formación de amiloide insoluble y su acumulación en placas estructuradas, en forma difusa en el neuropilo y en depósitos perivasculares – congofilia vascular- siempre que hayan sobrevivido en cautiverio o semilibertad, con cuidados especiales, el número de años suficiente para llegar a su “ancianidad” (76, 77). El porcentaje de simios ancianos con acumulación de amiloide varía grandemente según las especies (o las colonias de una especie estudiada: desde muy pocos individuos (<5%) hasta prácticamente todos, siendo, además, bastante controvertido el tipo y disposición de los depósitos, su relación con la amiloidosis humana en al EA y su relación con las alteraciones comportamentales propias de cada especie durante la fase senil.
Existen pocos estudios realizados de manera comparativa cognoscitiva/comportamental en vida y morfofuncional post-mortem para obtener conclusiones ya definitivas sobre envejecimiento normal y patológico, pero sí se tiene la certeza de que es un campo de estudio de gran relevancia para ello (76, 77, 79-81).
En algunos Macacus rhesus o Macacus fascicularis - macaco cangrejero o de cola larga-, estudiado por nuestro equipo de investigación, alrededor del 30% de los individuos que sobrepasan los 20 años de edad, presentan un síndrome cerebral neurodegenerativo muy acusado, similar a la EA humana, con cambios comportamentales muy profundos y gran acumulación de amiloide, frente al 70% del resto de individuos seniles con muy poco marcadas alteraciones neuropatológicas y déficits leves cognoscitivo-comportamentales (76, 77, 79) (Figura 11). Sin embargo, en otras especies, como el Chlorocebus aethiops - cercopiteco verde- también de la familia macaca, empleado en el estudio de posibles vacunas contra el Alzheimer (82, 83), todos los individuos que sobrepasaban los ocho años presentaban alteraciones neuropatológicas aunque con escasa repercusión cognoscitiva. Los estudios parecen mostrar la posible existencia de una transición entre la involución senil fisiológica y la patológica EA dentro de la cual ocurriría una fase estable sin demencia aun cuando estén presentes alteraciones neuropatológicas (77, 79).
En lo que respecta a la otra característica neuropatológica de la EA, la acumulación cerebral de agregados de proteína Tau en ovillos neurofibrilares y neuritas distróficas, especialmente la isoforma hiperfosforilizada, también hay que reseñar que solo la especie humana tiene un alto grado de estas alteraciones. En los primates no humanos aparecen alteraciones tau-dependientes en prosimios, pero luego desaparecen hasta llegar al hombre. En algún caso se han descrito algunas acumulaciones muy débiles tau-inmunopositivas en gorilas o chimpancés (79, 84) y, en nuestro caso, en macaco cangrejero (77, 79) (Figura 11). Si se consigue hacer un seguimiento cognoscitivo-comportamental, durante el tiempo necesario, de todos los individuos de una colonia de simios con suficiente población, se podría comparar la diferente involución senil cerebral en especies con posible patología amiloide y tau (humanos, prosimios), amiloide solamente (macacos) o sin patología amiloide o tau (titís).
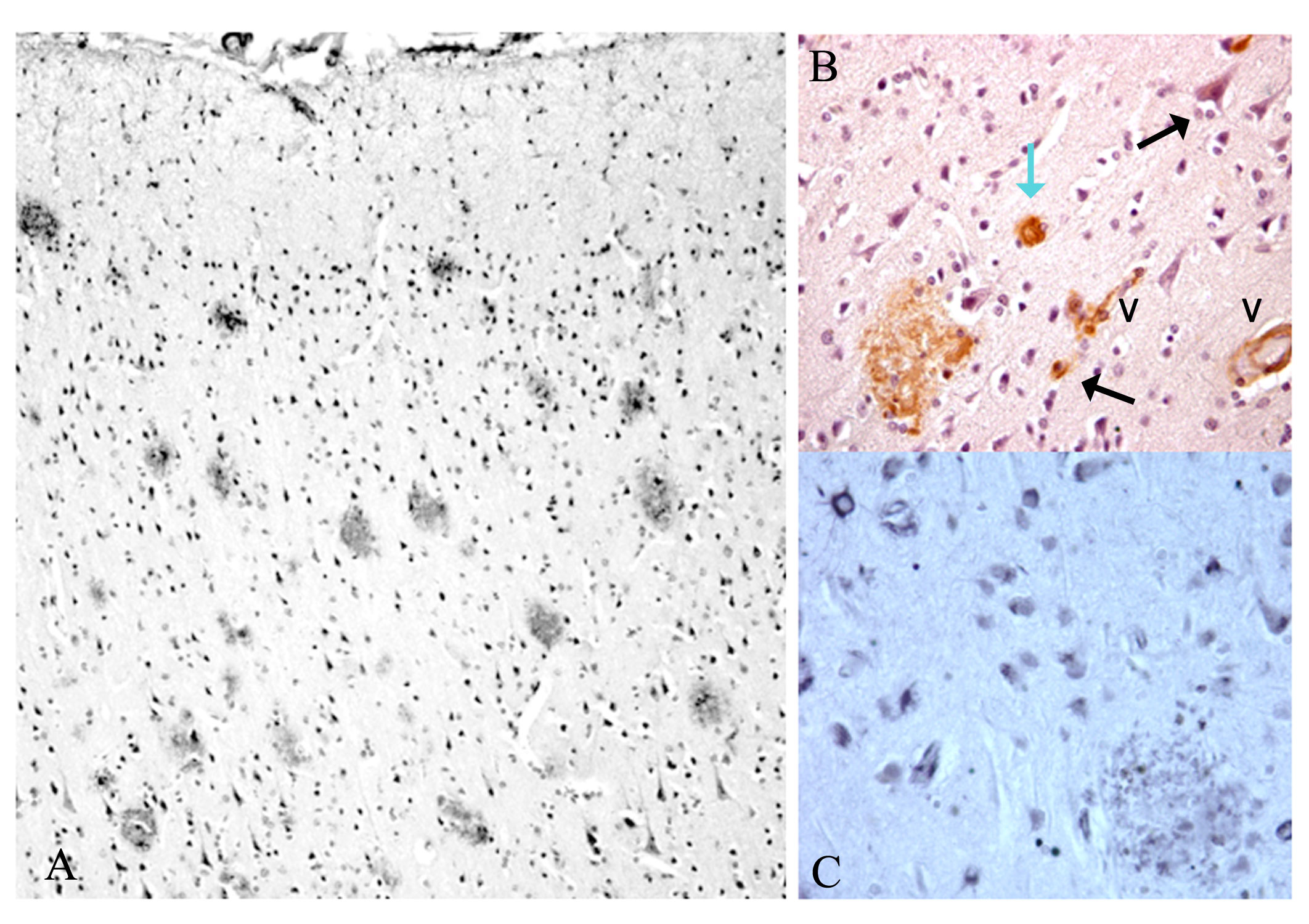
Figura 11.- Neuropatología Alzheimer en Macacusfasicularis (macaco congrejero o macaco de cola larga). Individuo de 36 años de edad con profunda involución comportamental. A. Corteza prefrontal con gran densidad de placas amiloideas (inmunotinción con anticuerpo 6E10 intensificado con Niquel). En B se observa además de una placa de tipo difuso, otros tipos de depósitos amiloideos en el neuropilo (como gruesas granulaciones o microplacas de tipo “burnout” del humano) (flecha en azul), granulaciones intracelulares en neuronas y células gliales (flechas en negro) y amiloideperivascular (congofiliopatía) (V). En C se observan granulaciones tau positivas (inmunotinción con anticuerpo clon tau-2) de distinto tamaño en neuronas y células gliales y en prolongaciones del neuropìlo o incluidas en una placa amiloidea (A=x200; B y C = x700).
e) Modelos transgénicos de Alzheimer experimental.
En los últimos años, la biotecnología ha propiciado el uso de animales genéticamente manipulados de tal forma que en campo de la EA podemos contar con animales experimentales que expresan isoformas aberrantes de proteínas implicadas (betaamiloide –Figura 12-, Tau) que inducen manifestaciones patológicas características de la senilidad patológica/EA. Diversos ratones transgénicos sencillos de APP (APP; APP+PSA) y Tau, o dobles y triples transgénicos (85-88) muestran como en la madurez adulta o en la senilidad se forman placas amiloideas o acumulaciones de proteína Tau. Muchos transgénicos tienen muy pocas alteraciones cognoscitivo-comportamentales, lo que ha servido para “desacreditar” estos modelos en el estudio de la senilidad patológica/EA, pero en otras es cierto que existe un mayor deterioro senil que en los controles normales (“wild type”) (87). Aunque estos ratones se han utilizado para estudios de patogenia y efectos de posibles fármacos o “vacunas” (88, 89) también nos brindan la oportunidad de estudiar las primeras fases de aparición de la patogenia y mecanismos implicados en la transición entre la “normalidad” y la “anormalidad”.
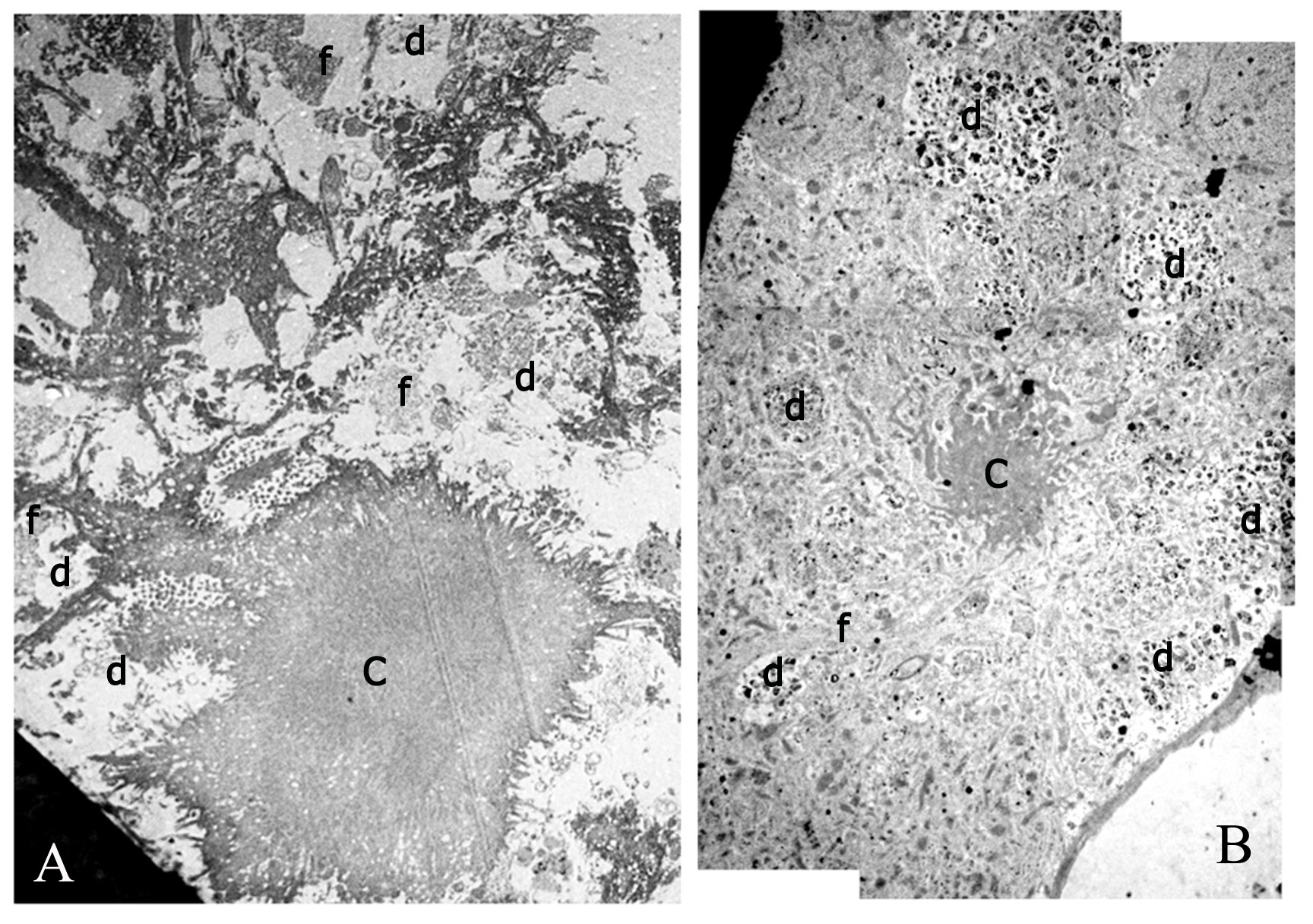
Figura 12.- Placas amiloideas observadas al Microscopio electrónico con core denso en humano (A) y en ratón trangénico APP+PS1. En ambos casos se observa el “core” central (c) formado por fibras densas de proteína beta amilideredeadas de dendritas distróficas (d) y material amilideo fibrilar difuso. Existen diferencias muy notables dehidasa : 1) la técnica de estudio, pues en el caso A se trata de tejido post-morten (3 horas), fijado por inmersión y la mayor parte de loas dendritas se han vascuolizado y destruido mientras se encuentran láminas ultadensas formadas por restos celulares neurovasales, gliales y aniloideas muy difíciles de estudio. Por el contrario en el ratón transgénico se observan las neuritas con grandes cantidades de detritus celulares rodeadas de las láminas de amiloide puramente fibrilar-granular. 2) El tipo de amiloide, la configuración del “core” y de la “corona”, las neuritas y las prolongaciones gliales tienen diferencias en las dos especies. (x6000).
6. Conclusiones
El problema teórico de si existe o no una doble vía de involución senil del cerebro, que podemos denominar, respectivamente, hacia la senilidad fisiológica o “normal” sin signos de demencia, o bien hacia la senilidad “patológica” o EA que se manifiesta por la aparición de una demencia progresiva, tiene también una muy importante repercusión práctica pues tanto la prevención como la asistencia de las personas mayores dependerán del posible número de individuos afectados: bien solamente los individuos de riesgo o bien toda la población. Los estudios ya realizados apuntan que existen características diferenciales muy marcadas entre los cerebros seniles más decantados hacia la normalidad o hacia la EA, pero no se conoce bien el estado real de las situaciones clínico-patológicas intermedias ni los mecanismos que propician el desarrollo de la patología. Existen algunos terrenos donde la investigación podría hacer avanzar nuestros conocimientos en el envejecimiento cerebral, especialmente en el estudio pormenorizado de los individuos controles sanos de entre 30 y 60 años, en el de los “centenarios” con y sin demencia y en grupos con características diferenciales neuropsicológicas que pudieran definirse en el transcurso de estudios epidemiológicos poblacionales (posibles estadíos intermedios de EA según el nuevo “lexicón” que se maneja en estos últimos años). También sería de gran interés estudiar comparativamente el envejecimiento cerebral humano con el de otras especies de mamíferos que no presentaran patología amiloide o tau y con los primates que pueden presentar patología amiloidea. En este último sentido, los animales transgénicos con patología inducida amiloide o/y tau también serían importantes fuentes de información.
7. referencias
1. Population Reference Bureau. Cuadro de datos de la población mundial 2012. http://www.prb.org/pdf12/2012-population-data-sheet_spanish.pdf
2. World Health Organization, Alzheimer’s Disease International. Dementia: a public health priority. WHO (OMS); Ginebra, 2012; p 12.
3. Plassman, B.L.; Langa, K.M.; Fisher, G.G.; Heeringa, S.G.; Weir, D.R.; Ofstedal, M.B.; Burke, J.R.; Hurd, M.D.; Potter, G.G.; Rodgers, W.L.; Steffens, D.C.; Willis, R.J.; Wallace, R.B. Prevalence of Dementia in the United States: The Aging, Demographics, and Memory Study. Neuroepidemiology 29, 125–132 (2007).
4. Thies, W.; Bleiler, L. Alzheimer's Association. Alzheimer's disease facts and figures. Alzheimer’s Dement, 9, 208-245 (2013).
5. Dubois, B.; Feldman, H. H,; Jacova, C.; Dekosky, S. T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; Meguro, K.; O'brien, J.; Pasquier, F.; Robert, P.; Rossor, M.; Salloway, S.; Stern, Y.; Visser, P. J. Scheltens P. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 6, 734-746 (2007).
6. Dubois, B.; Feldman, H. H.; Jacova, C.; Cummings, J. L.; Dekosky, S. T.; Barberger-Gateau, P.; Delacourte, A.; Frisoni, G.; Fox, N. C.; Galasko, D.; Gauthier, S.; Hampel, H.; Jicha, G. A.; Meguro, K.; O'Brien, J.; Pasquier, F.; Robert, P.; Rossor, M.; Salloway, S.; Sarazin, M.; de Souza, L. C.; Stern, Y.; Visser, P. J.; Scheltens, P. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol, 9, 1118-1127 (2010).
7. Lim YY, Maruff P, Pietrzak RH, Ellis KA, Darby D, Ames D, Harrington K, Martins RN, Masters CL, Szoeke C, Savage G, Villemagne VL, Rowe CC; AIBL Research Group. Aβ and cognitive change: Examining the preclinical and prodromal stages of Alzheimer's disease. Alzheimer´s Dement (en prensa) (Feb 28. doi: S1552-5260(13)02939-7) (2014).
8. Li, S.; Okonkwo, O.; Albert, M.; Wang, M. C. Variation in variables that predict progression from MCI to AD dementia over duration of follow-up. Am J Alzheimers Dis 2, 12-28 (2013).
9. Barnes, D.E.; Cenzer, I.S.; Yaffe, K.; Ritchie, C.S.; Lee, S. J. Alzheimer's Disease Neuroimaging Initiative; A point-based tool to predict conversion from mild cognitive impairment to probable Alzheimer's disease. Alzheimers Dement. 2014 (Feb 1. doi: S1552-5260(13)02941-5) (2014)
10 Castellani, R.J.; Perry, G. The complexities of the pathology-pathogenesis relationship in Alzheimer disease. Biochem Pharmacol (en prensa). doi: 10.1016/j.bcp.2014.01.009 (2014).
11. Mucke, L.; Selkoe, D. J. Neurotoxicity of amyloid β-protein: synaptic and network Dysfunction; Cold Spring Harb Perspect Med; 2(7):a006338. doi: 10.1101/cshperspect.a006338 (2012).
12. Swerdlow, R. H. Alzheimer's disease pathologic cascades: who comes first, what drives what. Neurotox Res 22, 182-194 (2012).
13. De Pedro-Cuesta, J.; Virués-Ortega, J.; Vega, S.; Seijo-Martínez, M.; Saz, P.; Rodríguez, F.; Rodríguez-Laso, A.; Reñé, R.; de las Heras, S. P.; Mateos, R.; Martínez-Martín, P.; Manubens, J. M.; Mahillo-Fernandez, I.; López-Pousa, S.; Lobo, A.; Reglà, J.L.; Gascón, J.; García, F. J.; Fernández-Martínez, M.; Boix, R.; Bermejo-Pareja, F.; Bergareche, A.; Benito-León, J.; de Arce, A.; del Barrio, J. L. 2009. Prevalence of dementia and major dementia subtypes in Spanish populations: a reanalysis of dementia prevalence surveys, 1990-2008. BMC Neurol. 9:55. doi: 10.1186/1471-2377-9-55.
14. Marín-Padilla, M. The Human Brain: Prenatal Development and Structure. Springer, Heidelberg, 2011.
15. Marín-Padilla, M. The mammalian neocortex new pyramidal neuron: a new conception. Front Neuroanat, 7, 51-63 (doi: 10.3389/fnana.2013.00051) (2014).
16. Ramón y Cajal, S. (1911). Histologie du systéme nerveux de l’homme et des vertebrés. Maloine, París, (1911) (reedición CSIC, Madrid, 1952).
17. Basak, O., Taylor, V. Stem cells of the adult mammalian brain and their niche. Cell. Mol Life Sci 66, 1057–1072 (2009).
18. Sala, C.; Segal, M. Dendritic spines: the locus of structural and functional plasticity. Physiol Rev 94(1), 141-88 (2014).
19. Vitureira, N.; Goda, Y. Cell biology in neuroscience: the interplay between Hebbian and homeostatic synaptic plasticity. J Cell Biol 203, 175-186 (2013)
20. Bhalla, U. S. Multiscale Modeling and Synaptic Plasticity. Prog Mol Biol Trans Sci 123, 351–386 (2014).
21. Toledano, A. Mecanismos de adaptación en el Sistema Nervioso. Su importancia en la patología y la terapéutica. Anal Real Acad Farm 65, 693-767 (1999).
22. Frol´kis, V.V.; Bezrukov, V.V. Aging of the central nervous system. Hum Physiol 4, 478-499 (1979).
23 Connor, J.R. jr.; Diamond, M.C.; Johnson A.R.E. Aging and Environmental Influences on two types of dendritic Spines in the rat occipital cortex. Experimental Neurology 70, 371-379 (1980)
24 Buell, S.; Coleman, P. Dendritic growth in the aged human brain and failure of growth in senile dementia. Science 206, 854-856 (1979).
25 Organización Mundial de la Salud. Clasificación Internacional de las Enfermedades, 10º edición. (CIE-10). Capítulo V. Trastornos mentales y del comportamiento. Descripciones clínicas y pautas para el diagnóstico. Meditor, Madrid, 1993, p 63.
26. Alzheimer, A. Über eine eigenartige erkrankung der hirnrinde. Allg Z Psychiat 64, 146-148 (1907).
27. Kraepelin, E . Psychiatrie: Ein Lehrbuch für Studierende und Ärzte. II. Band. Barth Verlag, Leipzig, 1910, p 21
28. American Psychiatric Association. Manual diagnóstico y estadístico de los trastornos mentales DSM IV. Masson, Barcelona, 2002, p 149.
29. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th Edition. DSM-5. ISBN 978-0-89042-554-1 (en prensa), 2015.
30. Khachaturian; ZS. Diagnosis of Alzheimer’s Disease. Arch Neurol 42, 1097-1105 (1985).
31 Mirra, S. S.; Heyman, A.; Mckeel, D.; Sumi, S. M.; Crain, B. J.; Brownlee, L. M.; Vogel, F. S.; Hughes, J. P.; Van Belle, G.; Berg, L. The Consortium to establish a registry for Alzheimer’s disease (CERAD); Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479-486 (1991).
32. Chételat, G.; Villemagne, V. L.; Pike, K. E.; Baron, J. C.; Bourgeat, P.; Jones, G.; Faux, N.G.; Ellis, K. A.; Salvado, O.; Szoeke, C.; Martins, R. N.; Ames, D.; Masters, C. L.; Rowe, C. C. Australian Imaging Biomarkers and Lifestyle Study of Ageing (AIBL) Research Group. Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain 133, 3349-3358 (2010).
33: Leuba, G.; Saini, K.; Zimmermann, V.; Giannakopoulos, P.; Bouras, C. Mild amyloid pathology in the primary visual system of nonagenarians and centenarians. Dement Geriatr Cogn Disord 12, 146-152 (2001).
34: Tolnay, M.; Calhoun, M.; Pham, H. C.; Egensperger, R.; Probst. A Low amyloid (Abeta) plaque load and relative predominance of diffuse plaques distinguish argyrophilic grain disease from Alzheimer's disease. Neuropathol Appl Neurobiol 25, 295-305 (1999).
35: Ince, P.; Irving, D.; MacArthur, F.; Perry, R. H. Quantitative neuropathological study of Alzheimer-type pathology in the hippocampus: comparison of senile dementia of Alzheimer type, senile dementia of Lewy body type, Parkinson's disease and non-demented elderly control patients. J Neurol Sci. 106,142-152 (1991).
36 Mesulam, M. M.; Weintraub, S.; Rogalski, E. J.; Wieneke, C.; Geula, C.; Bigio, E. H. Asymmetry and heterogeneity of Alzheimer's and frontotemporal pathology in primaryprogressive aphasia. Brain (en prensa) (2014).
37. Panza, F.; Frisardi, V.; Capurso, C.; D'Introno, A.; Colacicco, A. M.; Imbimbo, B. P.; Santamato, A.; Vendemiale, G.; Seripa, D.; Pilotto, A.; Capurso, A.; Solfrizzi, V.; Late-life depression, mild cognitive impairment, and dementia: possible continuum? Am J Geriatr Psychiatr 18, 98-116 (2010).
38. Wadsworth, L.P.; Lorius, N.; Donovan, N.J.; Locascio, J. J.; Rentz, D. M.; Johnson, K. A.; Sperling, R. A.; Marshall, G.A. Neuropsychiatric symptoms and global functional impairment along the Alzheimer's continuum. Dement Geriatr Cogn Disord 34, 96-111 (2012).
39. Zhang, M.; Wang, H.; Li, T.; Yu, X. Prevalence of neuropsychiatric symptoms across the declining memory continuum: an observational study in a memory clinic setting. Dement Geriatr Cogn Dis Extra 2, 200-208 (2012).
40. Rami, L.; Fortea, J.; Bosch, B.; Solé-Padullés, C.; Lladó, A.; Iranzo, A.; Sánchez-Valle, R.; Molinuevo, J. L. Cerebrospinal fluid biomarkers and memory present distinct associations along the continuum from healthy subjects to AD patients. J Alz Dis 23, 319-26 (2011).
41. Hyman, B. T.; Phelps, C. H.; Beach, T. G.; Bigio, E. H.; Cairns, N. J.; Carrillo, M. C.; Dickson, D. W.; Duyckaerts, C.; Frosch, M. P.; Masliah, E.; Mirra, S. S.; Nelson, P. T.; Schneider, J.A.; Thal, D.R.; Thies, B.; Trojanowski, J. Q.; Vinters, H.V., Montine, T.J. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. Dis. 8, 1-13 (2012).
42. Town, T.; Nikolic, V.; Tan, J. The microglial "activation" continuum: from innate to adaptive responses. J Neuroinflammation 31, 2-24 (2005).
43. Thal, D. R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Abeta deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791-1800 (2002).
44. Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239-259 (1991).
45. Nelson, P. T.; Alafuzoff, I.; Bigio, E. H.; Bouras, C.; Braakm, H.; Cairns, N. J:, Castellani, R. J.; Crain, B. J.; Davies, P.; Del Tredici, K.; Duyckaerts, C.; Frosch, M. P.; Haroutunian, V.; HofPR.; Hulette, C.M.; Hyman, B.T.; Iwatsubo, T.; Jellinger, K. A.; Jicha, G. A.; Kövari, E.; Kukull, W. A.; Leverenz, J. B.; Love, S.; Mackenzie, I. R.; Mann, D. M.; Masliah, E.; McKee, A. C.; Montine, T. J.; Morris, J. C.; Schneider, J. A.; Sonnen, J. A.; Thal, D. R.; Trojanowski, J. Q.; Troncoso, J. C.; Wisniewski, T.; Woltjer, R. L.; Beach, T. G. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71, 362-381 (2012).
46. Josephs, K. A.; Whitwell, J. L.; Ahmed, Z.; Shiung, M. M.; Weigand, S. D.; Knopman, D. S.; Boeve, B. F.; Parisi, J. E.; Petersen, R. C.; Dickson, D. W.; Jack, C.R.; Jr. Beta-amyloid burden is not associated with rates of brain atrophy. Ann Neurol 63, 204-12 (2008).
47 Toga, A. W. The clinical value of large neuroimaging data sets in Alzheimer's disease. Neuroimaging Clin N Am 22, 107-118 (2012).
48. Wirth, M.; Villeneuve, S.; Haase, C. M.; Madison, C. M.; Oh, H.; Landau, S. M.; Rabinovici, G. D.; Jagust, W. J. Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people. JAMA Neurol. 70,1512-1519 (2013)
49. Panza, F.; Frisardi, V.; Capurso, C.; D'Introno, A.; Colacicco, A. M.; Chiloiro, R.; Dellegrazie; F.; Di Palo, A.; Capurso, A.; Solfrizzi, V. Effect of donepezil on the continuum of depressive symptoms, mild cognitive impairment, and progression to dementia. J Am Geriatr Soc 58, 389-390 (2010).
50. Wright, L. K.; Litaker, M.; Laraia, M. T.; DeAndrade, S. Continuum of care for Alzheimer's disease: a nurse education and counseling program. Issues Ment Health Nurs 22, 231-252 (2001).
51. Cohen, A. D.; Rabinovici, G. D.; Mathis, C. A.; Jagust, W. J.; Klunk, W. E.; Ikonomovic, M. D. Using Pittsburgh Compound B for in vivo PET imaging of fibrillar amyloid-beta. Adv Pharmacol 64, 27-81 (2012).
52. Armstrong, R. A. The interface between Alzheimer's disease, normal aging, and related disorders. Curr Aging Sci 1, 122-132 (2008).
53. Saunders, A. M.; Strittmatter, W. J.; Schmechel, D.; George-Hyslop, P. H.; Pericak-Vance, M. A.; Joo, S. H.; Rosi, B. L.; Gusella, J. F.; Crapper-MacLachlan, D. R.; Alberts, M. J. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43, 1467-1472 (1993).
54. Ringman, J. M.; Coppola, G. New genes and new insights from old genes: update on Alzheimer disease. Continuum (Minneap Minn) 19, 358-71 (2013).
55. Serrano-Pozo, A.; Qian, J.; Monsell, S. E.; Frosch, M. P.; Betensky, R.A.; Hyman, B. T. Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. J Neuropathol Exp Neurol. 72,1182-1192 (2013).
56. Pappolla, M. A. La Neuropatología y la Biología Molecular de la Enfermedad de Alzheimer. En:. Neuropathología. Diagnóstico y Clínica. Cruz-Sánchez F.F., Ed.; Edimsa, Barcelona, 2000; p 543.
57. Penke, B.; Tóth, A. M.; Földi, I.; Szűcs, M.; Janáky, T. Intraneuronal β-amyloid and its interactions with proteins and subcellular organelles. Electrophoresis 33, 3608-3616 (2012).
58. Delacourte, A. General and dramatic glial reaction in Alzheimer brains. Neurology 40, 33-37 (1990)
59. Dickson; C., Vickers, J. C. The morphological phenotype of b-amyloid plaques and associated neuritic changes in Alzheimer's. Neuroscience 105, 99-107 (2001).
60. Wegiel, J.; Wang, K. C.; Tarnawski, M.; Lach, B. Microglia cells are the driving force in fibrillar plaque formation, whereas astrocytes are a leading factor in plague degradation. Acta Neuropathol 100, 356–364 (2000).
61. Bartus, R. T.; Dean, R. L.; 3rd, Beer, B. Lippa AS The cholinergic hypothesis of geriatric memory dysfunction. Science 30, 217(4558):408-14 (1982).
62. Mann, D. M.; Yates, P. O.; Marcyniuk, B. Alzheimer's presenile dementia, senile dementia of Alzheimer type and Down's syndrome in middle age form an age related continuum of pathological changes. Neuropathol Appl Neurobiol 10, 185-207 (1984).
63. Mecocci, P. Oxidative stress in mild cognitive impairment and Alzheimer disease: a continuum. J Alzheimers Dis. 2004 Apr;6(2):159-63.
64. Nixon, R. A.; Yang, D. S.; Lee, J. H. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4, 590-9 (2008).
65. Toledano, A.; Álvarez, MI.; Rivas, L.; Lacruz, C.; Martínez-Rodríguez, R. Amyloid precursor protein in the cerebellar cortex of Alzheimer 's disease patients devoid of cerebellar ß-amyloid deposits. Immunocytochemical study of five cases. J Neural Trans 106, 1151-1169 (1999).
66. Toledano, A. Hypotheses concerning the aetiology of the Alzheimer's disease. Pharmacopsychiat 21, 17-25 (1988).
67. Toledano, A.; Bentura, M. Pyritinol facilitates the recovery of cortical cholinergic deficits caused by nucleus basalis lesions J Neural Transm [P-D Section] 7, 195-209 (1994).
68. Drachman, D. A.; Leavitt, J. Human memory and the cholinergic system: relationship to aging? Arch Neurol 30, 113–121 (1974).
69. Coyle, J. T.; Price, D. L.; De Long, M. R. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science 219, 1184-1190 (1983).
70. Schneider, L. S. Alzheimer disease pharmacologic treatment and treatment research. Continuum (Minneap Minn). 19, 339-357 (2013).
71. Van Hoesen, G. W.; Augustinack, J. C.; Dierking, J.; Redman, S. J.; Thangavel, R. The parahippocampal gyrus in Alzheimer's disease. Clinical and preclinical neuroanatomical correlates. Ann N Y Acad Sci 911, 254-274 (2000).
72. de Toledo-Morrell, L.; Goncharova, I.; Dickerson, B.; Wilson, R. S.; Bennett, D. A. From healthy aging to early Alzheimer's disease: in vivo detection of entorhinal cortex atrophy. Ann N Y Acad Sci 911, 240-53 (2000).
73. Stricker, N. H.;(1), Chang, Y. L.; Fennema-Notestine, C.; Delano-Wood, L.; Salmon, D. P.; Bondi, M. W.; Dale, A. M. Alzheimer's Disease Neuroimaging Initiative. Distinct profiles of brain and cognitive changes in the very old with Alzheimer disease. Neurology 77, 713-21 (2011).
74. Villain, N.; Chételat, G.; Grassiot, B.; Bourgeat, P.; Jones, G.; Ellis, K. A.; Ames, D.; Martins, R. N.; Eustache, F.; Salvado, O.; Masters, C. L.; Rowe, C. C.; Villemagne, V. L. AIBL Research Group. Regional dynamics of amyloid-β deposition in healthy elderly, mild cognitive impairment and Alzheimer's disease: a voxelwise PiB-PET longitudinal study. Brain 135, 2126-2139 (2012).
75. Okello, A.; Koivunen, J.; Edison, P.; Archer, H. A.; Turkheimer, F. E.; Någren, K.; Bullock, R., Walker, Z.; Kennedy, A.; Fox, N. C.; Rossor, M. N.; Rinne, J. O.; Brooks, D. J. Conversion ofamyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology 73, 754-760 (2009).
76. Toledano, A.; Álvarez, M. I.; López-Rodríguez, A. B.; Toledano-Díaz, A.; Fernández-Verdecia, C. I. Does Alzheimer's disease exist in all primates? Alzheimer pathology in non-human primates and its pathophysiological implications (I). Neurologia 27, 354-69 (2012) [también versión en español]
77. Toledano, A.; Álvarez, M. I.; López-Rodríguez, A. B.; Toledano-Díaz, A.; Fernández-Verdecia, C. I. Does Alzheimer's disease exist in all primates? Alzheimer pathology in non-human primates and its pathophysiological implications (II) Neurologia. 29, 42-55 (2014) [también versión en español]
78. Hardy, J.; Selkoe, D. J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 19, 353-356 (2002).
79. Toledano, A.; Álvarez, M. I.; Carmona, P.; Toledano-Díaz, A.; Fernández-Verdecia, C. I. Alzheimer pathology in non-human primates and its pathophysiological implications. En: Primates. Classification, evolution and behavior. Hughes E. F.; Hill M. Eds. Nova Sciences Pub, New York, 2012; p 71-110.
80. Bons, N.; Rieger, F.; Prudhomme, D.; Fisher, A.; Krause, K. H. Microcebus murinus: a useful primate model for human cerebral aging and Alzheimer’s disease? Genes Brain Behav 5, 120-130 (2006).
81. Voytko, M. L.; Tinker, G. P. Cognitive function and its neural mechanism in nonhuman primate models of aging Alzheimer’s diseases, and menopause. Front Biosci 9, 1899-1914 (2004).
82. McGuire, M. T. The St Kitts vervet (Cercopithecus aethiops). J Med Primatol 3, 285-297 (1974).
83. Lemere, C. A.; Beierschmitt, A.; Iglesias, M.; Spooner, E. T.; Bloom, J.K.; Leverone, J. F. Alzheimer’s disease abeta vaccine reduces central nervous system abeta levels in a non-human primate, the Caribbean vervet. Am J Pathol 165, 83-97 (2004).
84. Rosen, R. F.; Farberg, A. S.; Gearing, M.; Dooyema, J.; Long, P. M.; Anderson, D. C. Tauopathy with paired helical filaments in an aged chimpanzee. J Comp Neurol 509, 259-270 (2008).
85. LaFerla, F. M.; Green, K. N. Animal models of Alzheimer disease. Cold Spring Harb Perspect Med, 2 (11), a006320. doi: 10.1101/cshperspect.a006320 (2012).
86. Scheffler, K.; Stenzel, J.; Krohn, M.; Lange, C.; Hofrichter, J.; Schumacher, T.; Brüning, T.; Plath, A. S.; Walker, L.; Pahnke, J. Determination of spatial and temporal distribution of microglia by 230nm-high-resolution, high-throughput automated analysis reveals different amyloid plaque populations in an APP/PS1 mouse model of Alzheimer's disease. Curr Alzheimer Res 8,781-788 (2011).
87. Bales, K.R. The value and limitations of transgenic mouse models used in drug discovery for Alzheimer's disease: an update. Expert Opin Drug Discov. 7, 281-297 (2012).
88. Howlett, D. R. APP transgenic mice and their application to drug discovery. Histol Histopathol 26, 1611-1632 (2011).
89. Boche, D.; Denham, N.; Holmes, C.; Nicoll, J. A. Neuropathology after active Abeta42 immunotherapy: implications for Alzheimer's disease pathogenesis. Acta Neuropathol 120, 369-384 (2010).
90. Fjell, A.; McEvoy, M.; Holland, L.; D.; Dale, A. M.; Walhovda, K. B. The Alzheimer’s disease Neuroimaging Initiative. What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog Neurobiol (en prensa) (2014). http://dx.doi.org/10.1016/j.pneurobio.2014.02.004.
91. Fischer, F. U.; Wolf, D.; Scheurich, A.; Fellgiebel, A. Association of structural global brain network properties with intelligence in normal aging. PLoS One 22, 9 (2014). doi: 10.1371/journal.pone.0086258.
92. Roussotte, F. F.; Gutman, B. A.; Madsen, S. K.; Colby, J. B.; Narr, K. L.; Thompson, P. M.; Alzheimer's disease Neuroimaging Initiative (ADNI). The apolipoprotein E epsilon 4 allele is associated with ventricular expansion rate and surface morphology in dementia and normal aging. Neurobiol Aging 35, 1309-1317 (2014).
93. Mosconi, L. Glucose metabolism in normal aging and Alzheimer's disease: Methodological and physiological considerations for PET studies. Clin Trans Imaging 1, doi: 10.1007/s40336-013-0026-y (2013)..
94. Nugent, S.; Tremblay, S.; Chen, K. W.; Ayutyanont, N.; Roontiva, A.; Castellano, C. A.; Fortier, M.; Roy, M.; Courchesne-Loyer, A.; Bocti, C.; Lepage, M.; Turcotte, E.; Fulop, T.; Reiman, E. M.; Cunnane, S. C. Brain glucose and acetoacetate metabolism: a comparison of young and older adults. Neurobiol Aging 35, 1386-1395 (2014).
95. Kövari, E.; Herrmann, F. R.; Bouras, C.; Gold, G. Amyloid deposition is decreasing in aging brains: an autopsy study of 1,599 older people. Neurology 28, 326-331 (2014)
96. Da, X.; Toledo, J. B.; Zee, J.; Wolk, D. A.; Xie, S. X.; Ou, Y.; Shacklett, A.; Parmpi, P.; Shaw, L.; Trojanowski, J. Q.; Davatzikos, C. Alzheimer's Neuroimaging Initiative. Integration and relative value of biomarkers for prediction of MCI to AD progression: Spatial patterns of brain atrophy, cognitive scores, APOE genotype and CSF biomarkers. Neuroimage Clin. 28, 164-173 (2013)
97. Madhyastha, T. M.; Grabowski, T. J. Age-related differences in the dynamic architecture of intrinsic networks. Brain Connect 30, (en prensa) (2014).
98. Toledano, A.; Álvarez M. I.; Toledano-Díaz,, A. Multiple neuronal and glial mechanisms can be involved in the response to the activation of nicotinic receptors. A. In: Horizons in Neuroscience Research. Volume 3 Neural Transmissions: Characteristics, Mechanisms and Malfunctioning, Costa, A. Villalba, E. (Eds). Nova Science Publishers, Inc.; Hauppauge, NY, pp 1-34 (2011).
99. Wolf, D.; Grothe, M.; Fischer, F. U.; Heinsen, H.; Kilimann, I.; Teipel, S.; Fellgiebel, A. Association of basal forebrain volumes and cognition in normal aging. Neuropsychologia 53, 54-63 (2014).
100. Wirth, M.; Villeneuve, S.; Haase, C. M.; Madison, C. M.; Oh, H.; Landau, S. M.; Rabinovici, G. D.; Jagust, W. J. Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people. JAMA Neurol 70, 1512-1519 (2013).
101. Jack, C.R Jr.; Wiste, H. J.; Weigand, S. D.; Knopman, D. S.; Lowe, V.; Vemuri, P.; Mielke, M. M.; Jones, D.T.; Senjem, M. L.; Gunter, J. L.; Gregg, B. E.; Pankratz, V. S.; Petersen, R. C. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology 12, 1732-1740 (2013).
102. Gold, P. E.; Newman, L. A.; Scavuzzo, C. J,; Korol, D. L. Modulation of multiple memory systems: from neurotransmitters to metabolic substrates. Hippocampus 23, 1053-1065 (2013).
103. Negash, S.; Wilson, R. S.; Leurgans, S. E.; Wolk, D. A.; Schneider, J. A.; Buchman, A. S.; Bennett, D. A.; Arnold, S. E. Resilient brain aging: characterization of discordance between Alzheimer's disease pathology and cognition. Curr Alzheimer Res 10, 844-851 (2013).
104. Shulman, J. M.; Chen, K.; Keenan, B. T.; Chibnik, L. B.; Fleisher, A.; Thiyyagura, P.; Roontiva, A.; McCabe, C.; Patsopoulos, N. A.; Corneveaux, J. J.; Yu, L.; Huentelman, M. J.; Evans, D. A.; Schneider, J. A.; Reiman, E. M.; De Jager, P. L.; Bennett, D.A. Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol 1, 1150-1157 (2013).
105. Cha, J.; Jo, H. J.; Kim, H. J.; Seo, S. W.; Kim, H. S.; Yoon, U., Park, H:, Na, D. L.; Lee, J. M. Functional alteration patterns of default mode networks: comparisons of normal aging, amnestic mild cognitive impairment and Alzheimer's disease. Eur J Neurosci 37, 1916-1924 (2013).
106. Hall, C. N.; Reynell, C.; Gesslein, B.; Hamilton, N. B.; Mishra, A.; Sutherland, B. A.; O'Farrell, F. M.; Buchan, A. M.; Lauritzen, M., Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 26, (en prensa) (2014) doi: 10.1038/nature13165.
107. Verkhratsky, A.; Rodríguez, J. J.; Parpura, V. Neuroglia in ageing and disease. Cell Tissue Res 21 (en prensa) (2014). doi: 10.1007/s00441-014-1814-z
108. Kilimann, I.; Grothe, M.; Heinsen, H.; Alho, E. J.; Grinberg, L.; Amaro, Jr. E.; Dos Santos, G. A.; da Silva, R. E.; Mitchell, A. J.; Frisoni, G. B.; Bokde, A. L.; Fellgiebel, A.; Filippi, M.; Hampel, H.; Klöppel, S.; Teipel, S. J. Subregional Basal Forebrain Atrophy in Alzheimer's Disease: A Multicenter Study. J Alzheimers Dis 6 (2014). doi:10.3233/JAD-132345
109. Scheff, S. W.; Neltner, J. H.; Nelson, P. T. Is synaptic loss a unique hallmark of Alzheimer's disease? Biochem Pharmacol. 9 (en prensa) (2014). doi: 10.1016/j.bcp.2013.12.028.
110. Lee, C. W.; Shih, Y. H.; Kuo, Y. M. Cerebrovascular pathology and amyloid plaque formation in Alzheimer's disease. Curr Alzheimer Res 11, 4-10 (2014).
111. Beason-Held, L. L.; Goh, J. O.; An, Y.; Kraut, M. A.; O'Brien, R. J.; Ferrucci, L.; Resnick, S. M. Changes in brain function occur years before the onset of cognitive impairment. J Neurosci 13, 18008-18014 (2013).
112. Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H. G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta. (en prensa) (2013) doi: 10.1016/j.bbadis.2013.10.015.
113. Jellinger, K. A.; Attems, J. Neuropathological approaches to cerebral aging and neuroplasticity. Dialogues Clin Neurosci 15, 29-43 (2013).
114. Lefort, R.; Pozueta, J.; Shelanski, M. Cross-linking of cell surface amyloid precursor protein leads to increased β-amyloid peptide production in hippocampal neurons: implications for Alzheimer's disease. J Neurosci 32, 10674-10685 (2012).
115. Shulman, J. M.; Chibnik, L. B.; Aubin, C.; Schneider, J. A.; Bennett, D.A.; De Jager, P. L. Intermediate phenotypes identify divergent pathways to Alzheimer's disease. PLoS One 5(6), e11244 (2010). doi: 10.1371/journal.pone.0011244.
116. Oikawa, N.; Kimura, N.; Yanagisawa, K. Alzheimer-type tau pathology in advanced aged nonhuman primate brains harboring substantial amyloid deposition. Brain Res 1315, 137-149 (2010).
117. Bell, K. F.; Ducatenzeiler, A.; Ribeiro-da-Silva, A.; Duff, K.; Bennett, D. A., Cuello, A. C. The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol Aging 27, 1644-1657 (2006).
118. Arnold, S.E.; Han, L. Y.; Clark, C. M.; Grossman, M.; Trojanowski, J. Q. Quantitative neurohistological features of frontotemporal degeneration. Neurobiol Aging 21, 913-919 (2000).
119. Toledano, A.; Álvarez M. I. Lesion-induced vertebrate models of Alzheimer Dementia. En Animal models of Dementia; Deyn, P.P.; Van Dam, D. (Eds). Humana Press; Amsterdam, 210; pp 295-345.
120. Toledano, A.; Álvarez M. I.; Toledano-Díaz,, A. Diversity and variability of the nicotine effects on different brain cortical regions. Therapeutic and toxicologic implications. CNS Agents M10, 180-206 (2010).