REVISIÓN |
Papel del tejido adiposo blanco, marrón y perivascular en las complicaciones vasculares asociadas a la obesidad
Almudena Gómez-Hernández1,2,3, Liliana Perdomo1,2 , Óscar Escribano1,2, Manuel Benito1,2
1Departamento de Bioquímica y Biología Molecular, Facultad de Farmacia, Universidad Complutense de Madrid, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos de Madrid (IdISSC), España. 2CIBER de Diabetes Enfermedades Metabólicas relacionadas, España
3Corresponding author: Almudena Gómez-Hernández, Departamento de Bioquímica y Biología Molecular, Facultad de Farmacia, Universidad Complutense de Madrid, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos de Madrid (IdISSC), España, Madrid-28040, Spain. Phone: 34-91-3941853; Fax: 34-91-3941779
e-mail: almudenagomez@farm.ucm.es
An. Real Acad. Farm. Vol. 80, Nº 2 (2014), pag. 322-346
resumen
En esta revisión se valora la contribución del tejido adiposo blanco, marrón y perivascular a la fisiopatología de las complicaciones metabólicas y vasculares asociadas a la obesidad. Para combatir la obesidad y evitar las crecientes complicaciones metabólicas y vasculares, además de los tratamientos establecidos, hay que avanzar en el conocimiento del tejido adiposo marrón y su prometedor potencial terapéutico. Dada la capacidad del tejido adiposo marrón en el gasto energético y los efectos sobre el metabolismo lipídico y glucídico, así como su potencial resistencia a la inflamación junto con el tejido adiposo perivascular; las nuevas perspectivas del tratamiento de la obesidad podrían centrarse en el diseño de nuevos fármacos o distintos regímenes o terapias que incrementen la cantidad y función del tejido adiposo marrón no sólo para luchar contra la obesidad sino también para prevenir la diabetes tipo 2 y otros desórdenes metabólicos y vasculares asociados a la misma. |
Palabras clave: Obesidad; Tejido adiposo marrón; Tejido adiposo blanco; Complicaciones vasculares.
abstract
This review analyzes the contribution of white, brown and perivascular adipose tissues to the pathophysiology of metabolic and vascular complications associated to obesity. To combat obesity and prevent its metabolic and vascular complications of high prevalence, in addition to conventional treatments a new insight in our knowledge of role of brown adipose tissue thermogenic function and its promising therapeutic potential in humans is much needed. Owing to the impact of brown adipose tissue on energy expenditure related to lipid and glucose metabolisms, as well as its potential resistance against inflammation together to perivascular adipose tissue, new perspectives in the obesity treatment might be focused in the design of new drugs, or different regimens or therapies, that increase the amount and activity of brown adipose tissue. These new treatments not only may contribute to combat obesity, but also prevent complications such as type 2 diabetes and other associated metabolic and vascular alterations. |
Keywords: Obesity; Brown adipose tissue; White adipose tissue; Vascular complications.
1. INTRODUCcióN
La obesidad es una enfermedad crónica de origen multifactorial que ha aumentado de forma considerable en los países desarrollados en las últimas décadas, siendo en la actualidad una epidemia mundial (1). La Organización Mundial de la Salud (OMS) considera obesos a aquellas personas que tengan un índice de masa corporal (IMC) superior a 30 kg/m2 y ha estimado que en el 2015 habrá 2300 millones de adultos con sobrepeso y 700 millones de obesos. Además está aumentando de forma preocupante la obesidad infantil y así en 2010, alrededor de 40 millones de niños menores de cinco años tenían sobrepeso (2). La obesidad se ha convertido en un problema de salud pública no sólo por el aumento de la estigmatización social, el problema económico que supone o el fallo en la calidad de vida, sino también por el riesgo asociado que presentan dichos pacientes a desarrollar otras patologías como la diabetes tipo 2, dislipidemias, hígado graso, aterosclerosis, enfermedad cardiovascular, enfermedad de Alzheimer, enfermedades óseas y con frecuencia algunos tipos de cánceres especialmente digestivos. Esto ocurre, al menos en parte, como resultado de la resistencia a la insulina inducida por la obesidad y el hecho de que el tejido adiposo no sólo sirve como reservorio de energía sino como un órgano endocrino secretor de hormonas, citoquinas y proteínas que afectan a la funcionalidad de las células y tejidos a lo largo de todo el organismo (3).
La obesidad se acompaña de un riesgo cardiovascular elevado por la coexistencia de otros factores de riesgo, particularmente dislipemia, hipertensión, insulinorresistencia y diabetes (4). De hecho, estos factores de riesgo están íntimamente ligados a un exceso de tejido adiposo, y más específicamente a una particular distribución corporal del mismo. Esta forma de distribución de la grasa en el obeso sí está claramente relacionada de manera independiente con la morbimortalidad cardiovascular a través de un síndrome metabólico aterogénico (5). Éste es el motivo por el que adquiere especial trascendencia clínica la medida, no sólo de la cuantía total de la grasa corporal, sino de su distribución, habida cuenta que tal distribución tiene más impacto en el riesgo cardiovascular que la obesidad por sí misma.
Una de las alteraciones metabólicas más deletéreas de la obesidad es la dislipemia que con frecuencia la acompaña y resulta altamente aterogénica (6). Su patrón fenotípico habitual es la hipertrigliceridemia preprandial, la hiperlipidemia no HDL postprandial, el aumento real en la producción de partículas densas y pequeñas de LDL, así como la baja producción de colesterol HDL.
En mamíferos el tejido adiposo está compuesto al menos por dos tipos muy distintas de grasas como son el tejido adiposo blanco (WAT) y el tejido adiposo marrón o pardo (BAT) que presentan diferencias en cuanto a su morfología, distribución, genes y función. El tejido adiposo blanco es el principal reservorio de energía y libera un gran número de hormonas y citoquinas que modulan el metabolismo del organismo y la resistencia a la insulina (7-10). El desarrollo de la obesidad no sólo depende del balance entre la ingesta y el gasto energético sino también del balance entre el tejido adiposo blanco, como principal reservorio energético, y el tejido adiposo marrón, especializado en el gasto energético en forma de termogénesis a través de la proteína mitocondrial desacoplante 1 (UCP-1). Además el BAT podría afectar al metabolismo del organismo y alterar la sensibilidad a la insulina (11, 12) al igual que modificar la susceptibilidad a desarrollar obesidad (13).
2. Papel del Tejido Adiposo Blanco en la Obesidad y sus Complicaciones Metabólicas y Vasculares Asociadas
2.1. Morfología y distribución del WAT. Implicación en el riesgo cardiovascular
El adipocito del WAT tiene una forma variable, aunque clásicamente es esférica de tamaño entre 25-200 μm. Además, tiene un núcleo periférico y plano y un citoplasma delgado que contiene una única gota lipídica grande que ocupa el 90% del volumen. Presenta escasas mitocondrias y un pequeño retículo endoplasmático liso y rugoso. WAT se compone de los adipocitos que se mantienen unidos por un tejido conectivo laxo que está adecuadamente vascularizado e inervado (14). Además de los adipocitos, WAT contiene macrófagos, leucocitos, fibroblastos, adipocitos, las células progenitoras y células endoteliales. La presencia de los fibroblastos, los macrófagos, y otros leucocitos, junto con los adipocitos, da cuenta de la gran variedad de proteínas que son secretadas por WAT bajo condiciones variables. El tejido adiposo blanco está distribuido a lo largo de todo el organismo y tiene diferentes compartimentos que varían en cuanto al tamaño celular del adipocito (15, 16) a la actividad metabólica y a su papel potencial en la resistencia a la insulina y otras complicaciones vasculares asociadas a la obesidad (17, 18). En humanos, se diferencian dos depósitos principales de tejido adiposo blanco: el depósito subcutáneo correspondiente al tejido adiposo que se localiza bajo la piel, y el depósito visceral. Hay dos tipos de tejido adiposo visceral: el mesentérico y el omental (19). El primero se encuentra envolviendo al intestino, el segundo se extiende desde la parte inferior del estómago, recubriendo el abdomen y es el que normalmente se emplea en el estudio de la grasa visceral. Hace tiempo que se sabe que el tejido adiposo visceral y el subcutáneo presentan numerosas diferencias anatómicas, celulares y moleculares (20, 21); por ejemplo, la irrigación de ambos tejidos es diferente (22), y los niveles de RNA mensajero (mRNA) de leptina en el tejido adiposo subcutáneo están incrementados respecto del visceral (20). Estos tejidos también son diferentes en cuanto a la capacidad de movilización de ácidos grasos, la grasa omental es más sensible a los efectos lipolíticos de las catecolaminas y menos sensible a los efectos antilipolíticos de la insulina; por tanto, este tejido tiene una mayor capacidad de movilización de ácidos grasos que el depósito subcutáneo (22, 23). Adicionalmente hay descritas numerosas diferencias entre el tejido adiposo visceral y subcutáneo referentes a la secreción de adipoquinas (24). En este sentido, una obesidad periférica se caracteriza por una acumulación de tejido adiposo subcutáneo y es más frecuente en mujeres. Este tipo de obesidad no se ha asociado a un mayor riesgo de sufrir patologías asociadas (25). Sin embargo, la obesidad central o abdominal es más frecuente en hombres y consiste en una acumulación de tejido adiposo visceral. Este tipo de obesidad se ha asociado, mediante estudios epidemiológicos, con un mayor riesgo de sufrir enfermedades tales como resistencia a la insulina, diabetes de tipo II e hipertensión, aumentando considerablemente el riesgo cardiovascular (26).
2.2. El WAT como reservorio de energía
En primer lugar, el tejido adiposo blanco es un órgano que constituye el mayor reservorio energético del organismo. La energía es almacenada en los adipocitos en forma de triglicéridos. La principal fuente de triglicéridos procede de los quilomicrones y las proteínas de muy baja densidad (VLDL) circulantes. En los humanos, el almacenamiento de los ácidos grasos en el tejido adiposo depende prácticamente de la liberación de los mismos desde las lipoproteínas por acción de la lipoproteína lipasa (LPL) (27). Tal es el protagonismo de esta enzima en el metabolismo lipídico, que se describe una acción proaterogénica de la LPL, expresada por el macrófago, y una acción antiaterogénica de la LPL, expresada en el tejido adiposo y músculo. Por tanto, esta enzima estaría implicada en las alteraciones lipídicas de la obesidad (28). Su actividad aumenta en el período pospandrial y se inhibe en el ayuno y está incrementada en el tejido adiposo tanto de humanos como de animales de experimentación obesos (29-34). Sin embargo, la capacidad de respuesta de la LPL a la insulina y a la alimentación en pacientes obesos está disminuida (32, 35).
Otro de los procesos metabólicos que se producen en el tejido adiposo es la lipólisis, donde los triglicéridos almacenados en el tejido adiposo son hidrolizados a ácidos grasos y glicerol. El paso limitante de la lipólisis está controlado por la lipasa sensible a hormonas (HSL). Dicha hormona presenta una intensa regulación. Así, la activación de los receptores β adrenérgicos produce un aumento de los niveles intracelulares de AMPc y estimula la fosforilación activadora (P-Ser 552), catalizada por la proteína quinasa activada por AMPc (PKA) de la HSL (36). Sin embargo, la activación de los receptores α2 adrenérgicos, favorece la reducción de los niveles intracelulares de AMPc, produciendo una menor activación de PKA y por tanto, de HSL. Así, las catecolaminas tienen un efecto dual sobre la lipólisis, y su efecto neto depende del balance entre los receptores α2 y β adrenérgicos. Otra de las hormonas inhibidoras de la lipólisis, es la insulina, que induce la activación de PI3K y de la fosfodiesterasa III, que a su vez produce la inactivación de AMPc. En adipocitos de pacientes obesos, la lipólisis basal está aumentada y falla la lipólisis estimulada por catecolamina además de existir un descenso en la expresión de HSL (37). Se ha descrito que alteraciones en la lipólisis inducidas por catecolaminas puede tener un papel importante en el desarrollo de la obesidad en humanos, así como sus complicaciones metabólicas y vasculares asociadas. La capacidad lipolítica parece tener un componente hereditario, aunque también puede verse afectada por el propio sobrepeso y por el grado de actividad física (38). Asimismo, se ha descrito defectos de la HSL en familias de obesos y polimorfismos de los genes para HSL y para los receptores adrenérgicos β2 y β3 en asociación a la obesidad humana. Los adipocitos de pacientes obesos se caracterizan por presentar niveles altos de receptores adrenérgicos α2 y un ratio elevado de receptores adrenérgicos α2/β (39). Además, en modelos animales se ha demostrado una correlación positiva entre el grado de obesidad y el ratio de receptores adrenérgicos α2/β (40).
2.3. El WAT como órgano endocrino
El tejido adiposo blanco no sólo es un órgano de reservorio de energía sino también un órgano secretor de ciertas moléculas que tienen una acción endocrina, paracrina y autocrina (41). Algunas de estas moléculas secretadas por los adipocitos están implicadas en la regulación del peso corporal (leptina, adiponectina), en la respuesta inflamatoria generada localmente en una situación de obesidad (TNF-a, IL-6 e IL-1β), en la función vascular (Ang II y PAI-1) o reproductora (estrógenos, entre otras).
La leptina es una hormona secretada principalmente por los adipocitos teniendo un papel relevante en la regulación del peso corporal a través de sus efectos centrales, sobre el apetito y periféricos, sobre el gasto energético (42). La concentración de leptina circulante disminuye en condiciones de ayuno o restricción calórica y aumenta en respuesta a la ingesta, principalmente en respuesta a glucosa (43, 44). Sin embargo, la gran mayoría de pacientes obesos presentan concentraciones elevadas de leptina y están aumentados en relación al grado de adiposidad y de hiperinsulinemia, lo que ha llevado al concepto de leptinorresistencia (45). Esta hiperleptinemia ha sido involucrada en la insulinorresistencia del obeso a través de alteraciones en la fosforilación del receptor de la insulina (46). Otra de las hormonas secretadas por los adipocitos que participa en el control de la ingesta es la adiponectina (Acrp30/AdipoQ). Es una adipocitoquina implicada en la regulación del metabolismo energético del organismo, ya que estimula la oxidación de ácidos grasos, reduce los triglicéridos plasmáticos y mejora el metabolismo de la glucosa mediante un aumento de la sensibilidad a la insulina (47). En diversos estudios, se ha observado hipoadiponectinemia en pacientes con obesidad, diabetes mellitus y arteriopatía coronaria (48, 49). Además de sus propiedades antidiabetogénicas, la adiponectina posee un efecto antiaterogénico y también tiene una relación inversa con otros factores de riesgo como la presión arterial, el colesterol total y las lipoproteínas de baja densidad (LDL) (50-53). Los estudios transversales de población muestran que concentraciones bajas de adiponectina están relacionadas con un aumento del perfil de riesgo metabólico y cardiovascular (54, 55). Recientemente, se ha identificado una nueva molécula, la resistina, secretada por los adipocitos maduros y que podría ser el nexo de unión entre la obesidad y el desarrollo de resistencia a la insulina (56). En roedores parece estar clara su implicación en la resistencia a la insulina. Sus niveles circulantes se incrementan durante la obesidad, su bloqueo mejora la homeostasis de la glucosa y su administración ejerce un efecto negativo sobre los tejidos diana de la insulina. En humanos, sin embargo, el papel de la resistina no está ni mucho menos esclarecido, y los trabajos publicados son bastante contradictorios. Parece que esta hormona podría ejercer algún papel en la respuesta inflamatoria debido a su mayoritaria expresión en células mononucleares (57). Se requieren, por tanto, nuevos estudios para determinar el papel de esta molécula tanto en la obesidad como en la resistencia a la insulina.
Diversas citoquinas proinflamatorias son secretadas por distintos tipos celulares incluidos los adipocitos. Tienen una acción paracrina o autocrina en el propio tejido y participan en la respuesta inflamatoria local que se produce en los adipocitos de pacientes obesos. Se ha descrito que los niveles de TNF-a en el adipocito están correlacionados positivamente con el tamaño de los depósitos adiposos. Además, la expresión del RNA mensajero del TNF-α está aumentada en el tejido adiposo de distintos modelos murinos de obesidad y diabetes y de pacientes obesos, relacionándose dicho aumento con el desarrollo de resistencia a la insulina (58, 59). Por un lado, el TNF-a activa la lipólisis e inhibe los niveles de LPL y GLUT-4 como un mecanismo que trata de reducir el tamaño excesivo de los depósitos grasos. Sin embargo, los niveles altos de TNF-a en el tejido adiposo podría deberse a alguna de las alteraciones metabólicas asociadas a la obesidad como es la resistencia a la insulina. Así, el TNF-a aumenta los niveles de ácidos grasos libres, reduciendo la sensibilidad a insulina, y tiene un efecto inhibidor directo en la acción de la insulina en el hígado, incrementando la producción de glucosa hepática (60, 61). Sin embargo, las acciones biológicas del TNF-α podría estar influenciadas por la expresión de dos receptores: p60 TNFR (TNF-R1) y p80 TNFR (TNF-R2). La expresión de TNF-R1 está positivamente correlacionada con el índice de masa corporal y el tamaño de los adipocitos, mientras que la expresión de TNF-R2 muestra asociaciones positivas con la concentración circulante de insulina y triglicéridos (62). Así, interfiriendo la vía de señalización del TNF-α se protege la resistencia a la insulina inducida por obesidad (63). La neutralización del TNF-α usando un anticuerpo monoclonal reduce los niveles de glucosa del modelo murino diabético KKAy (64) y mejora el control glicémico en pacientes con diabetes tipo 2 (65). Del mismo modo, el tratamiento con un anticuerpo anti-TNF-α durante seis semanas redujo la hiperglucemia en el ayuno, la intolerancia a la glucosa y mejoró la sensibilidad a la insulina en el tejido adiposo blanco visceral, principalmente en el compartimento gonadal, del ratón BATIRKO de 52 semanas, que presenta un aumento de la adiposidad asociado a una lipoatrofia marrón severa. En este modelo, el tratamiento con anti-TNF-a redujo la activación de NF-kB en ambos tejidos adiposos, y la expresión de moléculas controladas por dicho factor de transcripción tanto en el tejido adiposo blanco gonadal, en el tejido adiposo marrón así como en la aorta. Además, las alteraciones vasculares, como la resistencia a la insulina vascular, y disfunción vascular fueron revertidas por el tratamiento con anti-TNF-a (66).
El angiotensinógeno y el inhibidor del activador del plasminógeno (PAI-1) son moléculas secretadas también por los adipocitos y cuya expresión génica está aumentada en situaciones de obesidad (67, 68), teniendo un efecto deletéreo sobre la función vascular. Además, otro componente del sistema renina-angiotensina que también tiene el adipocito es la angiotensina II, que posee un efecto estimulante sobre la diferenciación del tejido adiposo y tiene su implicación en la regulación de la adiposidad debido a sus acciones lipogénicas (69). En relación a la secreción de PAI-1 por el tejido adiposo, se ha observado una mayor producción en la grasa visceral que en la grasa subcutánea. En este sentido, los niveles de PAI-1 estaban aumentados en la obesidad central relacionándose con las alteraciones vasculares asociados a la misma (68).
2.4. Papel del WAT en la inflamación generada en la obesidad y en las alteraciones vasculares asociadas
En una situación de obesidad asociada a una resistencia a la insulina, tanto el exceso de dieta como la propia obesidad producen una acumulación de lípidos en los adipocitos, iniciándose un estrés en la célula y la activación de la vía de las JNK y del factor nuclear de transcripción kB (NF-kB) (70, 71). Estas vías de señalización inflamatorias regulan la fosforilación de proteínas y distintos eventos celulares transcripcionales que conducen a un aumento por parte del adipocito en la producción de moléculas proinflamatorias, incluidas el TNF-a, IL-6, leptina y resistina, de quimioquinas tales como la proteína quimioatractante de monocitos (MCP-1), y de otros mediadores proaterogénicos, como por ejemplo PAI-1. Moléculas de adhesión endotelial (ej.: ICAM-1 y VCAM-1), y moléculas quimioatractantes (ej.: CCX) se unen a integrinas y receptores de quimioquinas (CCR), respectivamente, y favorecen el reclutamiento de monocitos y otras células inflamatorias al tejido adiposo. Los monocitos una vez en el interior se diferencian a macrófagos y amplifican la respuesta inflamatoria produciendo muchas de las mismas citoquinas y quimioquinas inflamatorias mencionadas previamente (72) (Figura 1). Un número reciente de artículos han sugerido también que los linfocitos T podrían desempeñar un papel importante en la producción de citoquinas proinflamatorias y en el reclutamiento de macrófagos al tejido adiposo en obesos. Al igual que los monocitos, las células T que circulan a través del organismo, infiltran los tejidos periféricos en respuesta a las señales de quimioquinas y citoquinas. El infiltrado de linfocitos precede a la población de monocitos en respuesta a dieta grasa y podría proporcionar mediadores proinflamatorios que promueven el reclutamiento y la activación de macrófagos (Figura 1). Los linfocitos T citotóxicos del linaje CD8 están muy enriquecidos en el tejido adiposo de ratones sometidos a dieta grasa, resultando consistente con el aumento significativo de células CD8 en pacientes obesos (73). Así, ratones deficientes en CD8 fueron parcialmente resistentes a desarrollar obesidad inducida por dieta rica en grasas, mientras que la transferencia de células CD8 agravaba la inflamación del tejido adiposo (73).
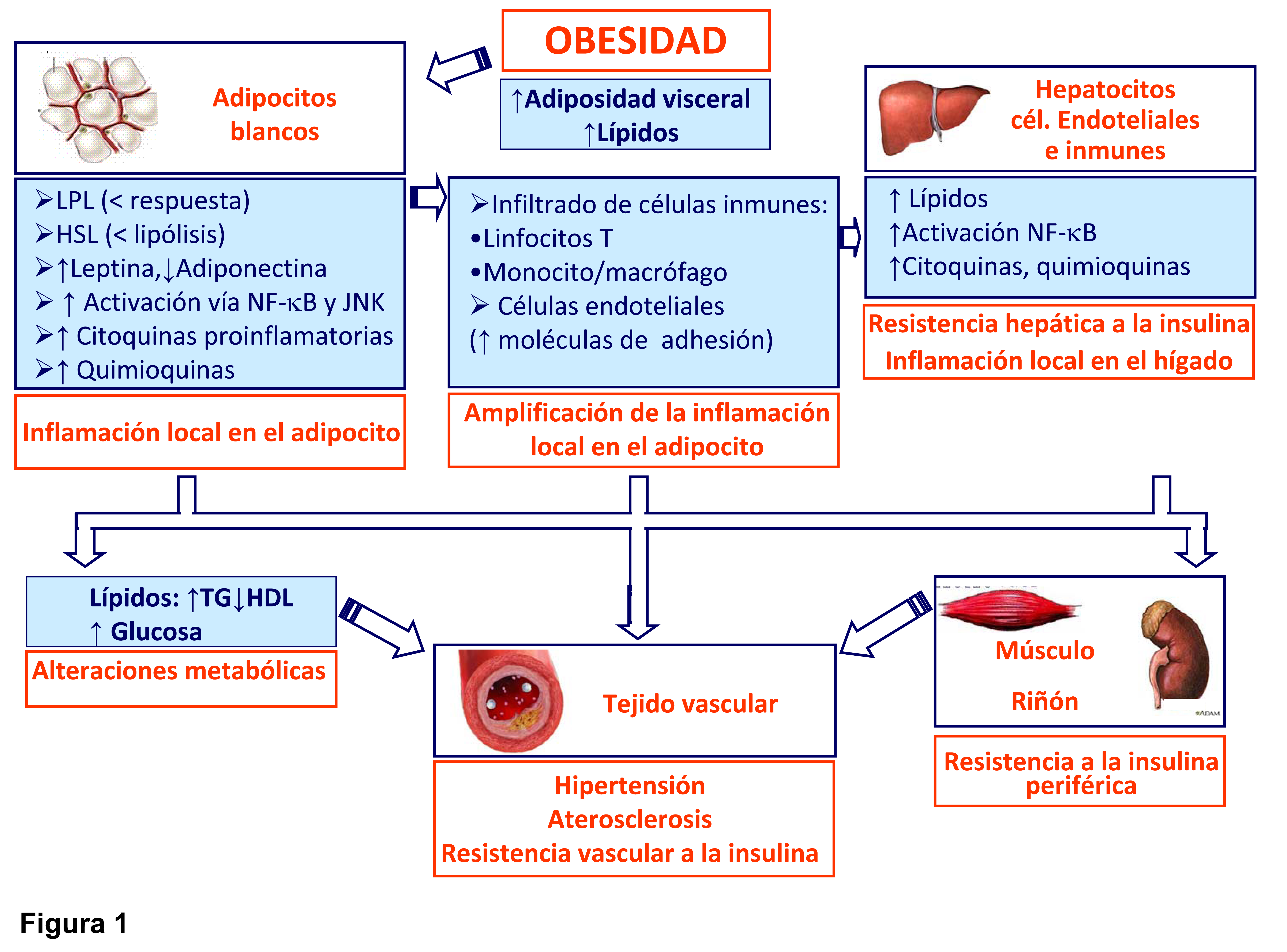
Figura 1.- Contribución del tejido adiposo blanco en la obesidad y las complicaciones metabólicas y vasculares asociadas. En una situación de obesidad se produce una acumulación de lípidos en los adipocitos, iniciándose un estrés en la célula y la activación de la vía de las JNK y del NF-kB, generándose una inflamación local en el adipocito blanco. Ésta puede exportarse a través de la vía portal al hígado y finalmente, a otros tejidos periféricos, como el territorio vascular donde podría producir aterosclerosis, hipertensión y resistencia vascular a la insulina.
Además de los propios adipocitos y las células inflamatorias, otros tipos celulares podría participar en dicha respuesta inflamatoria. Así, el tejido adiposo está vascularizado, con múltiples capilares en contacto con cada adipocito (74). En este sentido, para la expansión de la grasa, la microcirculación podría desempeñar un papel clave en la inflamación del tejido adiposo. Así, en un endotelio normal que es antiadherente, los leucocitos no se adhieren, mientras que después de la administración de dieta rica en grasas se ha demostrado que dicho endotelio expresa moléculas de adhesión y une leucocitos (75). Las células endoteliales del tejido adiposo podrían aumentar la expresión de proteínas de adhesión, como ICAM-1, VCAM-1, E-selectina o P-selectina en respuesta a un aumento de la adiposidad y favorecer así la adhesión de células inflamatorias como linfocitos T y monocitos (76).
El aumento de la adiposidad no sólo activa la respuesta inflamatoria en el adipocito sino también a través de la vía portal en el hígado (77) (Figura 1). Esto sugiere que la acumulación lipídica en los hepatocitos o esteatosis podría inducir una respuesta inflamatoria subaguda en el hígado, que es similar a la inflamación en el tejido adiposo que sigue a la acumulación lipídica en el adipocito. Las moléculas proinflamatorias producidas en la grasa abdominal a través de la circulación portal podrían ser las responsables del inicio de la inflamación hepática. Además en el propio hepatocito graso se produce la activación de NF-kB y un aumento de la expresión de citoquinas, incluidas el TNF-a, IL-6 e IL-1b (77). Las citoquinas proinflamatorias participan en el desarrollo de la resistencia a la insulina y activan a los macrófagos hepáticos residentes llamadas células Kupffer. El hígado está densamente poblado por células Kupffer que representan aproximadamente el 5% del total de las células. En la obesidad con el aumento de la adiposidad no aumenta el número de células Kupffer sino su activación (77). En el hígado, al igual que en el tejido adiposo hay distintos tipos de células que participan en el inflamación y en la resistencia a la insulina a nivel local del hepatocito como son las células inmunes y las células endoteliales (78). Por tanto, los mediadores proinflamatorios y proaterogénicos que son producidos por el tejido adiposo y el hígado y asociados a las células inmunes generan una inflamación sistémica que produce resistencia a la insulina en el músculo esquelético y otros tejidos periféricos. En el tejido vascular, además de producir resistencia a la insulina vascular, podrían contribuir a iniciar el proceso aterogénico (79) (Figura 1).
3. Papel del Tejido Adiposo Marrón en la Obesidad y sus Complicaciones Metabólicas y Vasculares Asociadas
3.1. Morfología y distribución del BAT
El tejido adiposo marrón está formado por adipocitos marrones y células progenitoras de adipocitos. Así, el adipocito marrón tiene una forma poligonal con un núcleo oval y centrado en un citoplasma grande que contiene múltiples y pequeñas gotas lipídicas. Posee un gran número de mitocondrias y un retículo endoplasmático poco desarrollado. Además, se encuentra altamente vascularizado e inervado (80). Originalmente, se pensó que el BAT estaba presente en los seres humanos sólo durante el período neonatal. Sin embargo, datos recientes han demostrado que los adultos conservan algunos depósitos metabólicamente activos de BAT que responden al frío y a la activación simpática del sistema nervioso. Dichos depósitos son UCP-1 positivos y son detectados por tomografía por emisión de positrones (PET). Actualmente, en humanos, el tejido adiposo marrón se ha descrito que está localizado en depósitos de la región cervical, supraclavicular, paravertebral, mediastinal, para-aórtica y suprarrenal (81). Además, también se han localizado pequeños grupos de adipocitos marrones en el interior del músculo esquelético en adultos humanos (13). Por otro lado, recientes datos han mostrado que los adipocitos marrones encontrados en el interior de otros depósitos de tejido adiposo blanco no son derivados del linaje myf5, como son los clásicos adipocitos marrones del tejido interescapular de los roedores, y se denominan células beige (82). Dichas células son positivas para UCP-1, con alta capacidad respiratoria, con características de tejido adiposo blanco y marrón y con una alta sensibilidad a la hormona polipeptídica irisina (83). En este sentido, se habría mostrado previamente que la irisina secretada por el músculo esquelético y que se incrementa con el ejercicio, induce la “marronización” del tejido adiposo blanco subcutáneo. Sin embargo, esta proteína tiene poco efecto en los clásicos adipocitos marrones aislados del depósito interescapular (84). Todo ello, sugiere que la respuesta a la irisina podría ser una característica selectiva de las células beige localizadas en el interior de los depósitos de tejido adiposo blanco subcutáneo.
El tejido adiposo marrón además de estar implicado en la termogénesis, recientes estudios han demostrado que pudiera estar implicado en la liberación de triglicéridos y en la utilización de glucosa, además de servir como fuente de adipoquinas y poseer distinta función inflamatoria comparada con el tejido adiposo blanco.
3.2. La relación de BAT con la obesidad y sus complicaciones vasculares y metabólicas
La activación del tejido adiposo marrón reduce la adiposidad y protege al ratón de la obesidad inducida por la dieta rica en grasas (85, 86). Así, la pérdida de la masa del tejido adiposo marrón (87), como puede ser una lipoatrofia marrón severa por una deleción del receptor de la insulina en ese tejido (66, 88), o la pérdida de UCP-1 (89) confiere susceptibilidad a desarrollar obesidad en ratones. En los últimos años, se ha descrito que la cantidad de BAT estaba inversamente correlacionada con el índice de masa corporal en humanos, especialmente en personas de avanzada edad (90). Además, se ha demostrado recientemente que el tejido adiposo marrón podría proteger frente a múltiples enfermedades relacionadas con el envejecimiento (14). Así, individuos con depósitos de BAT más pequeños son más susceptibles a acumular tejido adiposo blanco y aumentar su peso corporal y tener un mayor riesgo de desarrollar enfermedades metabólicas y vasculares asociadas a dicha obesidad (85, 91).
3.3. El BAT y la termogénesis
Una de la principales funciones del tejido adiposo marrón es la activación de la termogénesis a través de la activación de la proteína desacoplante 1 (UCP-1), generando calor a partir de los ácidos grasos (92). En la termogénesis, el tejido adiposo marrón utiliza, en primer lugar, los lípidos almacenados como sustrato para generar calor. Esta fase temprana de la termogénesis corresponde con la liberación de norepinefrina del sistema nervioso simpático activando la liberación de ácidos grasos de las gotas de triglicéridos. Algunos de estos ácidos grasos activan a UCP-1. Los ácidos grasos remanentes son importados a la mitocondria y allí se realiza la termogénesis con la disipación de energía en forma de calor debido a la acción de UCP-1. Como el tejido adiposo marrón es un pequeño porcentaje del total del peso corporal, para mantener una termogénesis sostenida es necesario importar y quemar triglicéridos circulantes (93).
3.4. El BAT y el metabolismo lipídico y glucídico
El tejido adiposo marrón además de estar implicado en la termogénesis, recientes trabajos han demostrado que podría tener un papel destacado en el metabolismo lipídico y glucídico (Figura 2). En primer lugar, el tejido adiposo marrón podría estar implicado en la eliminación de triglicéridos. Así, las lipoproteínas ricas en triglicéridos (TRLs) transportan lípidos en la circulación, donde una porción de los ácidos grasos puede ser liberada por la LPL (94). Otros órganos periféricos, como el tejido adiposo blanco y el músculo esquelético captan ácidos grasos, mientras que las partículas remanentes ricas en colesterol son eliminadas por el hígado (94). Además, niveles elevados de triglicéridos y de partículas remanentes ricas en colesterol, como ocurre en la dislipidemia diabética, representan factores de riesgo para desarrollar enfermedades cardiovasculares (95-97). Se ha descrito que el aumento de la actividad del tejido adiposo marrón por exposiciones cortas al frío podría controlar el metabolismo de las TRLs en ratón, regulando la eliminación de dichas lipoproteínas y el exceso de lípidos circulantes (93) y así disminuyendo los niveles de triglicéridos y aumentado ligeramente los niveles de HDL (Figura 2). Así, los ácidos grasos son eficientemente introducidos en el propio tejido adiposo marrón debido a un programa metabólico que empuja a las TRLs a una captación muy eficiente de los ácidos grasos. Este proceso asociado con un aumento de la expresión de VEGF (98), conduce a un incremento de la permeabilidad para las lipoproteínas, permitiendo que los triglicéridos salgan de los capilares. La activación del tejido adiposo marrón por norepinefrina no sólo activa la liberación de los ácidos grasos de los triglicéridos dentro del propio tejido adiposo marrón junto con una mayor producción de VEGF, también aumenta la expresión de la LPL (93, 99). Por tanto, LPL degrada los triglicéridos y permite que los ácidos grasos puedan estar disponibles a través de transportadores de membrana plasmática como el CD36. Además, se ha demostrado en humanos que la activación de BAT por exposición al frío, incrementa su metabolismo oxidativo, reduciendo el contenido de triglicéridos y contribuyendo de forma decisiva al gasto energético (100). Por tanto, la activación de BAT sería capaz de corregir las hiperlipidemias y mejorar los efectos deletéreos de la obesidad y la dislipidemia como pueden ser la resistencia a la insulina o el proceso aterogénico. Recientemente, se ha descrito que el tejido adiposo epicárdico (EAT) podría ser un depósito de tejido adiposo marrón activo que es capaz de modificar los niveles de lípidos circulantes alterados, así aumentado los niveles de HDL y disminuyendo los niveles de triglicéridos (101).
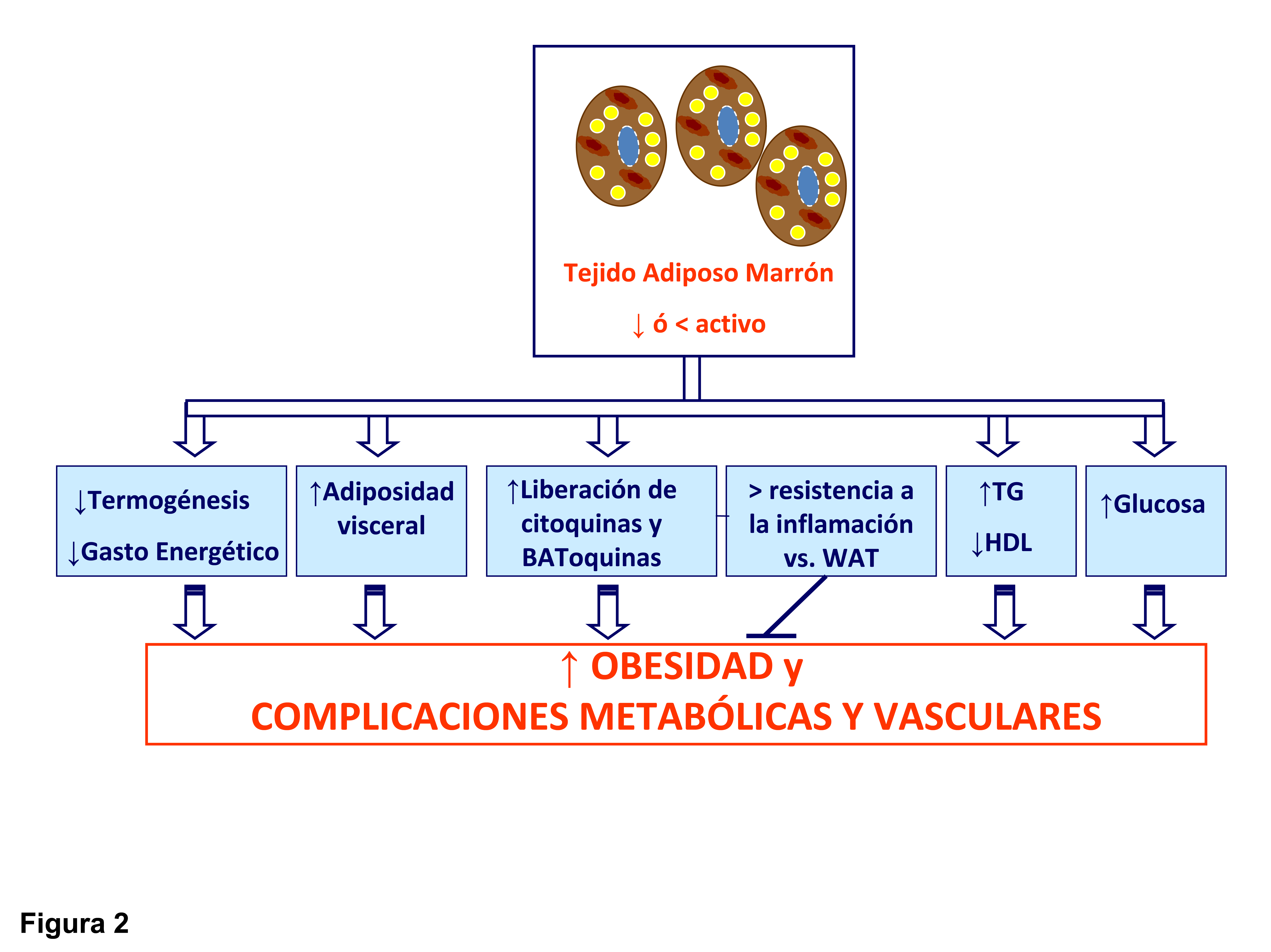
Figura 2.- Contribución del tejido adiposo marrón en la obesidad y las complicaciones metabólicas y vasculares asociadas. Una de las posibles causas que inducen el desarrollo de la obesidad podría ser una disminución de la cantidad y actividad del tejido adiposo marrón. En esta situación se produciría una alteración de funciones que realiza el tejido adiposo marrón en el metabolismo lipídico, glucídico así como el perfil de expresión de citoquinas y adipoquinas, favoreciéndose la obesidad y las complicaciones metabólicas y vasculares asociadas a la misma.
Por otro lado, también se ha descrito que el tejido adiposo marrón, podría también estar regulando el metabolismo glucídico (93). Dicho tejido tiene una alta captación de glucosa debido probablemente a una intensa combustión de glucosa en la mitocondria, más que por acción de la insulina. La mitocondria del tejido marrón utiliza el piruvato para la combustión siempre que UCP-1 esté activo por los ácidos grasos (102). Asimismo, los transportadores de glucosa, GLUT-1 y GLUT-4, podrían estar implicados en la captación de glucosa por parte del BAT ya que la actividad y expresión de ambos transportadores está aumentada tanto por frío como por norepinefrina (93, 103, 104).
3.5. El BAT como órgano endocrino
El tejido adiposo marrón es un órgano endocrino al igual que el tejido adiposo blanco. Secreta distintas citoquinas, hormonas y otros factores, como TNF-a, adiponectina y leptina al igual que el tejido adiposo blanco. Sin embargo, hay un gran número de moléculas que son exclusivamente secretadas por el tejido adiposo marrón, las llamadas BAToquinas (adipoquinas derivadas de BAT). Muchas de estas BAToquinas, incluida el factor de crecimiento fibroblástico (FGF21), son requeridas en la adaptación al frío y en la estimulación adrenérgica (105-107). Además, FGF21 puede actuar también de forma directa sobre el tejido adiposo marrón, independientemente del control adrenérgico, lo que permitirá abrir nuevas vías para explorar mecanismos de control de la grasa corporal (107). Otras proteínas secretadas también por el BAT, como adipsina, FGF2, IGF-1, prostaglandinas y la adenosina, desempeñan también funciones autocrinas.
Además, el tejido adiposo marrón secreta otros factores como IL-6 y factores neurotróficos, como el BDNF y el factor de crecimiento nervioso (NGF), los cuales podrían tener papeles únicos en el BAT con respecto al WAT (108-111). La secreción de NGF se produce principalmente por la proliferación de preadipocitos marrones, que promueve la inervación simpática del tejido, y a su vez permite una mayor estimulación por norepinefrina en los adipocitos marrones. Otros factores paracrinos, además de los neurotróficos, sintetizados por el BAT son el factor de crecimiento endotelial vascular (VEGF), el angiotensinógeno y el óxido nítrico. La expresión de VEGF con sus receptores, FLK-1 y FLK-4, es alta en la proliferación y madurez de adipocitos marrones, manteniendo alto el nivel de vascularización de este tejido. Tanto la noradrenalina como la exposición al frío inducen un aumento de la expresión de VEGF en BAT (112). Por otro lado, tanto la óxido nítrico inducible (iNOS) como la endotelial (eNOS) se expresan en el BAT y la noradrenalina induce un aumento en la producción de óxido nítrico (NO) que inhibe la oxidación mitocondrial y promueve un rápido aumento en el flujo sanguíneo (113).
A diferencia del tejido adiposo blanco que rápidamente es infiltrado por células inflamatorias en respuesta a la obesidad inducida por dieta grasa, el tejido adiposo marrón no parece acumular tanto infiltrado de macrófagos (114) (Figura 2). Esto puede deberse, a que el BAT tiene un mayor número de mitocondrias que le permite metabolizar los ácidos grasos a través de la b-oxidación. Sin embargo, en el WAT, la capacidad para metabolizar los lípidos estaría superada, teniendo efectos lipotóxicos y desencadenando la respuesta inflamatoria y facilitando el infiltrado de macrófagos y células inmunes. En este sentido, otro grupo reciente ha demostrado que los macrófagos del tejido adiposo marrón no tienen el mismo perfil de expresión de citoquinas y quimioquinas que los macrófagos del tejido adiposo blanco (115).
4. Papel del Tejido Adiposo Perivascular en la Obesidad y sus Complicaciones Metabólicas y Vasculares Asociadas
4.1. Morfología y distribución del PVAT
El tejido adiposo perivascular (PVAT) se encuentra rodeando la arteria coronaria (o tejido adiposo epicárdico), la aorta (tejido adiposo periaórtico) y otros vasos sistémicos así como el lecho microcirculatorio de las mesentéricas, músculo, riñón y tejido adiposo a excepción de la circulación cerebral (116). Se une al exterior de la capa adventicia sin ninguna estructura laminar o barrera organizada que separe a las dos. Dependiendo del lecho vascular, PVAT puede tener más o menos características de tejido adiposo blanco o marrón. Así, se ha descrito que el PVAT de la arteria abdominal sería prácticamente tejido adiposo blanco, el PVAT de las arterias coronarias humanas tendría un fenotipo intermedio entre adiposo blanco y marrón y el PVAT de la arteria aórtica torácica sería muy similar al tejido adiposo marrón (92, 117). La vascularización y la inervación del PVAT también varían de forma considerable con la localización y podría ayudar a explicar las distintas características funcionales de PVAT.
4.2. Papel de PVAT en la obesidad y en las complicaciones vasculares asociadas
PVAT está constituido por los adipocitos, fibroblastos, células madre, células que penetran el vaso vasorum y células inflamatorias infiltradas como macrófagos y linfocitos T, pudiendo ser estas últimas relevantes en determinadas situaciones patológicas (116).
El tejido adiposo perivascular que se extiende desde la capa adventicia es un modulador clave de la función vascular tanto en sujetos delgados como en animales delgados de experimentación. Sin embargo, en una situación de obesidad, el tejido perivascular aumenta su tamaño, creando un entorno de hipoxia que podría disminuir la producción de adiponectina que tiene efectos protectores frente a la aterogénesis y otras complicaciones vasculares (117) (Figura 3). Además, en la obesidad así como en el síndrome metabólico PVAT pierde su capacidad vasorreguladora porque hay una menor liberación de adipoquinas vasodilatoras y simultáneamente se liberan más factores que promueven la vasoconstricción (118). Así, el tejido adiposo perivascular tiene propiedades anticontráctiles y algunos estudios encuentran que éstas se pierden en la obesidad (116) (Figura 3). También se ha descrito, que el aumento de PVAT podría estar correlacionado positivamente con la cantidad de tejido adiposo intra-abdominal (119). Por tanto, en una situación de obesidad y aterosclerosis, el PVAT además de expandirse puede ser infiltrado por células inmunes, como macrófagos y linfocitos T (114, 121). La acumulación de linfocitos T favorecería la expansión del tejido adiposo debido a la estimulación de la adipogénesis por producción de 15d-PGJ2 y activación de PPAR-g (121). Sin embargo, los macrófagos no afectan a la expansión del PVAT, pero producen citoquinas que alteran la secreción de adipoquinas de dicho tejido. Así, la producción de leptina (123, 124), de citoquinas y quimioquinas proinflamatorias (125, 126) además de especies libres de oxígeno (116, 127) y ácidos grasos no esterificados (128) está aumentada en el PVAT de pacientes obesos y de animales de experimentación obesos (Figura 3).
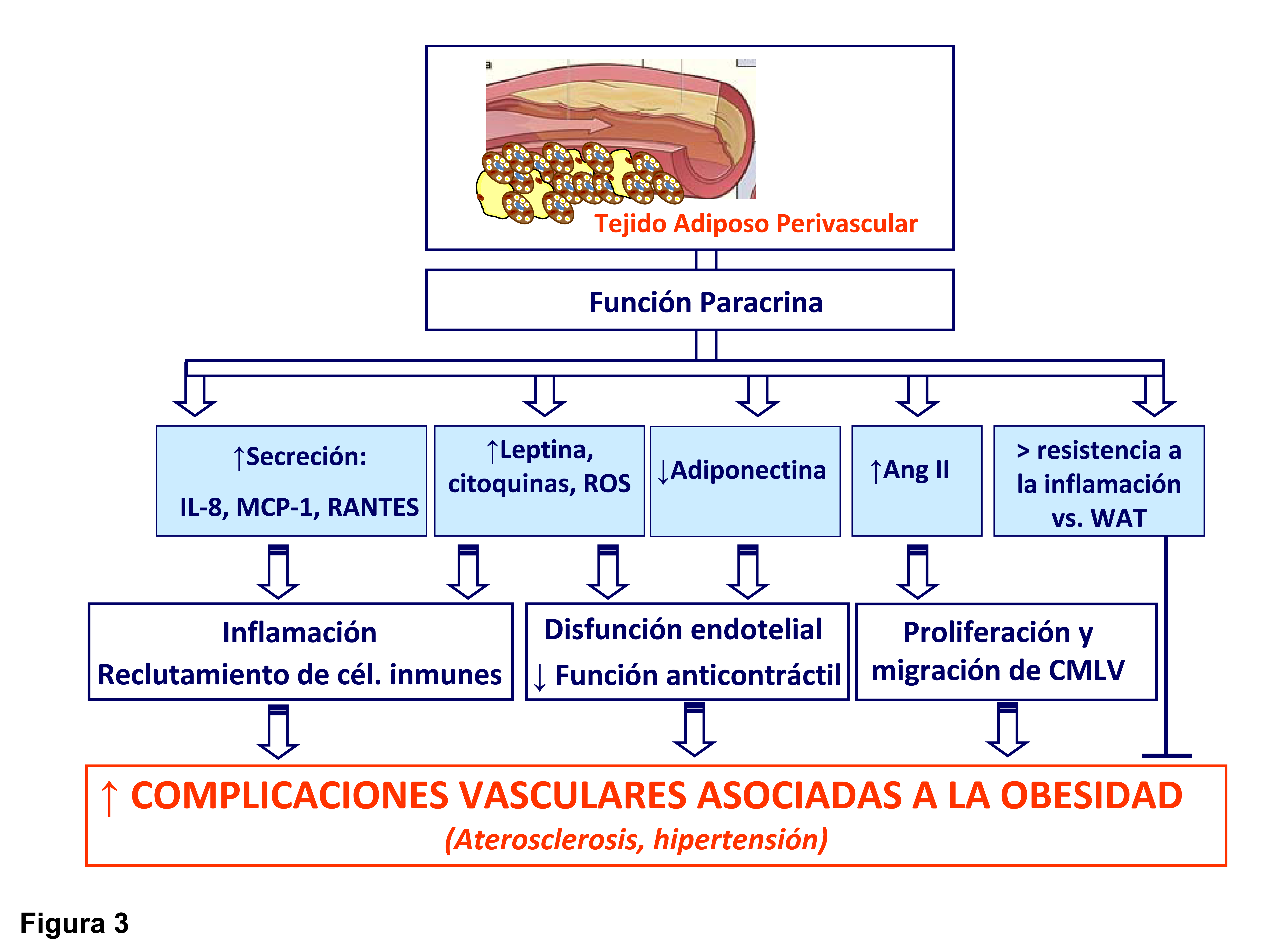
Figura 3.- Contribución del tejido adiposo perivascular en las complicaciones vasculares asociadas a la obesidad. El tejido adiposo perivascular interacciona con el endotelio, las células de músculo liso vascular y las células inmunes. Igualmente, se producen ciertos mediadores que estarían implicados en las posibles alteraciones vasculares asociadas a la obesidad, tales como la hipertensión y la aterogénesis.
Sin embargo, se ha descrito que las propiedades inflamatorias del tejido adiposo epicárdico son independientes de la obesidad (129). En este sentido, estudios recientes también han demostrado que el PVAT que está alrededor de la arteria aorta torácica es muy similar a BAT en cuanto a su morfología y el perfil de expresión genético en ratones (114). Además, el tejido adiposo perivascular de la aorta torácica junto con el BAT son más resistentes a la inflamación inducida por un dieta rica en grasas en un modelo murino (114) (Figura 3). Sería interesante, comprobar si el tejido adiposo perivascular en pacientes obesos con y sin enfermedad cardiovascular tiene una morfología y un perfil genético parecido también al BAT como ocurre en los modelos murinos estudiados. Así, podría resultar beneficiosa la activación del fenotipo tejido adiposo marrón en el tejido adiposo perivascular con el fin de prevenir enfermedades vasculares asociadas a la obesidad, como hipertensión y la aterogénesis.
5. Nuevas Perspectivas en el Tratamiento de la Obesidad
Las primeras indicaciones para el tratamiento de la obesidad junto con la restricción calórica es el ejercicio físico de manera dosificada y con cargas adecuadas a la condición física de cada paciente obeso. Existen considerables evidencias que la restricción calórica aumenta la esperanza de vida (130) y reduce el riesgo de desarrollar diabetes, enfermedad cardiovascular, desórdenes degenerativos y algunos tipos de cáncer (130, 131). Los dos mecanismos principales que estarían implicados en los efectos “anti-envejecimiento” y “anti-obesidad” de la restricción calórica, serían: (1) una menor producción de radicales libres mitocondriales (132), y (2) un aumento de la producción de proteínas resistentes al estrés celular (133). Además de la restricción calórica, hay evidencias que muestran que un balance energético mantenido durante varios meses, donde se incluye un aumento del gasto energético suele resultar efectivo en la disminución de la adiposidad. Esta reducción se produce principalmente en la grasa visceral, que es la que posee receptores y actividad lipolítica mayor que el tejido adiposo de otras regiones (134). Además, personas con un buen estado físico tienen la lipólisis en reposo mayor que los inactivos (135). Otro aspecto que mejora el ejercicio físico en pacientes obesos es el perfil lipídico. En primer lugar, eleva las HDL y por tanto disminuye el cociente LDL/HDL y el riesgo cardiovascular (136). Además, el ejercicio aumenta el tamaño de las partículas de LDL y HDL resultando un perfil lipídico menos aterogénico que las partículas pequeñas de LDL y HDL, propias de los pacientes obesos (136). Asimismo, un ejercicio físico regular también disminuye los niveles de triglicéridos en aquellos individuos con valores inicialmente altos, a través de una mejora en la sensibilidad a la insulina (137, 138). También el ejercicio físico produce distintas adaptaciones metabólicas que pueden resultar beneficiosas para el tratamiento de la obesidad. Se produce un aumento del potencial oxidativo y así, se favorece que se metabolicen más lípidos e hidratos de carbono de forma aeróbica, produciendo adaptaciones periféricas muy deseables. Por tanto, el ejercicio físico normaliza el perfil metabólico y permite la disminución de la morbimortalidad por estas causas (139, 140).
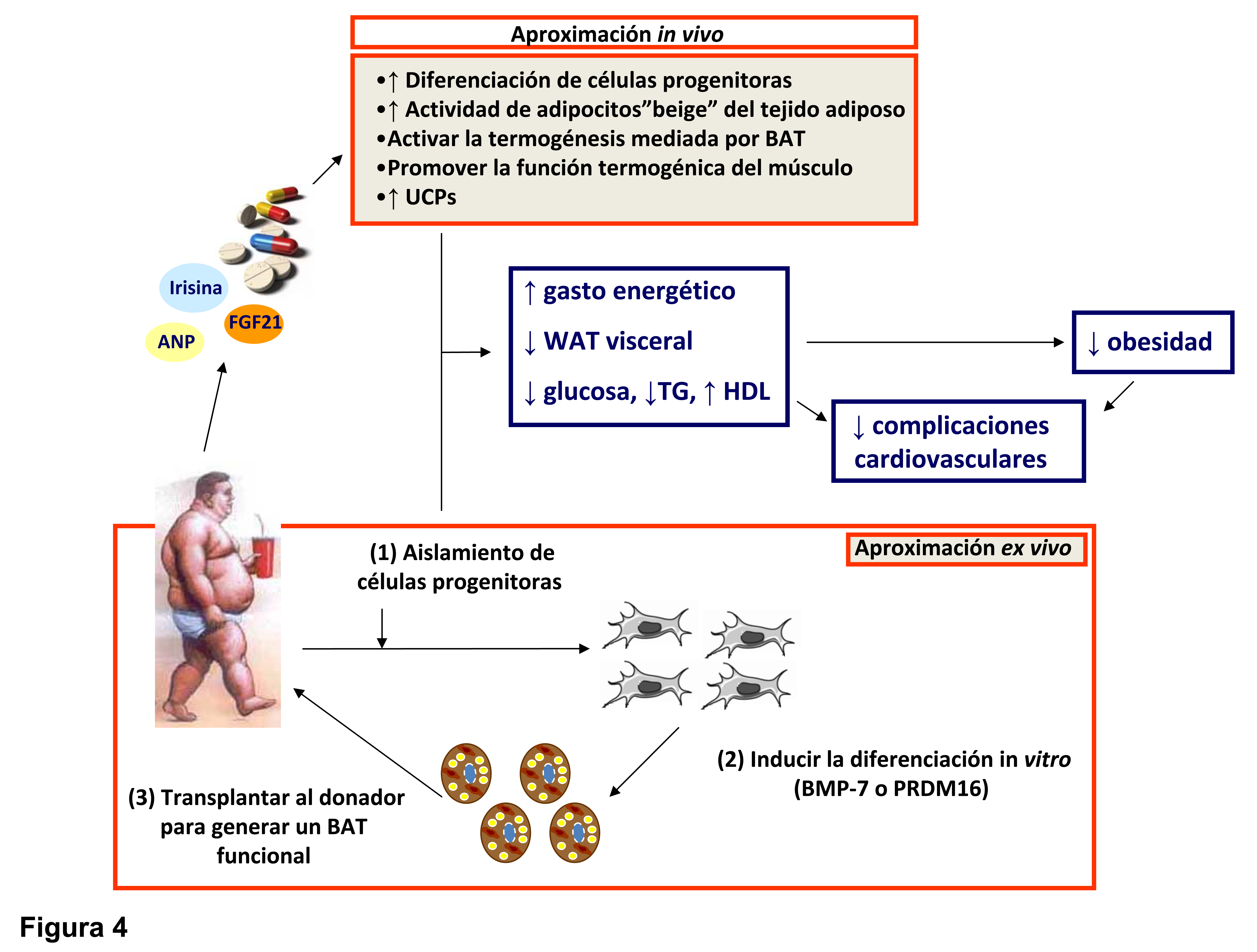
Figura 4.- Nuevas perspectivas en el tratamiento de la obesidad. Basado en los conocimientos actuales como futuras aproximaciones tanto in vivo como ex vivo para el tratamiento de la obesidad. Todas ellas están destinadas a producir la activación del tejido adiposo marrón o la diferenciación de células progenitoras o beiges en adipocitos marrones o promover la termogénesis en el músculo con el fin de favorecer la termogénesis, el gasto energético, la reducción de la adiposidad visceral así como un mejor control de la glucosa y el perfil lipídico, todo ello reduciendo la obesidad y las complicaciones vasculares asociadas.
Para poder combatir esta epidemia mundial que es la obesidad y evitar así las complicaciones metabólicas y vasculares que está continuamente creciendo, además de los tratamientos establecidos, tanto la restricción calórica, el ejercicio, los distintos fármacos o la cirugía, hay que aunar esfuerzos para avanzar en el conocimiento del tejido adiposo marrón y su prometedor potencial terapéutico frente a la obesidad y las complicaciones asociadas (141-143). Se ha descrito que la respuesta adaptativa del tejido adiposo marrón a un moderado e intermitente estrés a través de la activación simpática, podría aumentar la proliferación y diferenciación de células progenitoras de adipocitos marrones, además de incrementar la masa mitocondrial y la expresión de UCP-1 en tejido adiposo marrón (144). Todos estos efectos, junto con la estimulación de BAT en los depósitos de tejido adiposo blanco o en el músculo esquelético (145, 146), podrían aumentar el gasto energético y reducir el estrés oxidativo y la adiposidad visceral y en consecuencia, una mayor resistencia a desarrollar obesidad y enfermedades metabólicas y vasculares asociadas a la misma. Curiosamente, el trasplante de tejido adiposo marrón (0.1-0.4 g) a la cavidad visceral de un ratón es capaz de prevenir la ganancia de peso y mejorar la homeostasis glucídica en el ratón obeso sometido a dieta grasa (147). Por otro lado, como se han identificado que los depósitos de tejido adiposo marrón en humanos están compuestos por adipocitos beige (83); estos resultados podrían abrir nuevas vías de investigación para determinar si este tipo de células podrían tener cierto potencial terapéutico. Así, la irisina que es una molécula circulante endógena y media algunos beneficios que produce el ejercicio y además activa a los adipocitos beige en roedores, podría representar uno de los caminos aplicables a humanos. Finalmente, dada la capacidad del tejido adiposo marrón en el gasto energético y los efectos sobre el metabolismo lipídico y glucídico, así como su potencial resistencia a la inflamación junto con el tejido adiposo perivascular; las nuevas perspectivas del tratamiento de la obesidad podrían centrarse en el diseño de nuevos fármacos o distintos regímenes o terapias que incrementen la cantidad y función del tejido adiposo marrón no sólo para luchar contra la obesidad sino también para prevenir la diabetes tipo 2 y otros desórdenes metabólicos y vasculares (Figura 4).
6. bibliografía
1. Caballero B. The global epidemic of obesity: an overview. Epidemiol Rev. 2007;29:1-5.
2. Obesidad y sobrepeso. Nota descriptiva N°311. Oreganización Mundial de la Salud. Marzo de 2011
3. Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007; 131(2):242-56.
4. Cascio G, Schiera G, Di Liegro I. Dietary fatty acids in metabolic syndrome, diabetes and cardiovascular diseases. Curr Diabetes Rev. 2012; 8(1):2-17.
5. Mathieu P, Lemieux I, Després JP. Obesity, inflammation, and cardiovascular risk. Clin Pharmacol Ther. 2010; 87(4):407-16.
6. Grundy SM. Metabolic complications of obesity. Endocrine. 2000;13(2):155-65.
7. Aldhahi W, Hamdy O. Adipokines, inflammation, and the endothelium in diabetes. Curr Diab Rep. 2003; 3(4):293–8.
8. Ronti T, Lupattelli G, Mannarino E. The endocrine function of adipose tissue: an update. Clin Endocrinol (Oxf) 2006; 64(4):355–65.
9. Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006; 4(4):263–73.
10. Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006; 444 (7121):847–53.
11. Lowell BB, S-Susulic V, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, et al. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993; 366(6457):740–2.
12. Yang X, Enerbäck S, Smith U. Reduced expression of FOXC2 and brown adipogenic genes in human subjects with insulin resistance. Obes Res. 2003; 11 (10):1182–91.
13. Almind K, Manieri M, Sivitz WI, Cinti S, Kahn CR. Ectopic brown adipose tissue in muscle provides a mechanism for differences in risk of metabolic syndrome in mice. Proc Natl Acad Sci U S A. 2007; 104(7):2366–71.
14. Mattson MP. Perspective: Does brown fat protect against diseases of aging? Ageing Res Rev. 2010; 9(1):69-76.
15. Johnson PR, Zucker LM, Cruce JA, Hirsch J. Cellularity of adipose depots in the genetically obese Zucker rat. J Lipid Res. 1971; 12(6):706-14.
16. Krotkiewski M, Björntorp P, Sjöström L, Smith U. Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J Clin Invest. 1983; 72(3):1150-62.
17. Coon PJ, Rogus EM, Drinkwater D, Muller DC, Goldberg AP. Role of body fat distribution in the decline in insulin sensitivity and glucose tolerance with age. J Clin Endocrinol Metab. 1992; 75(4):1125-32.
18. Gastaldelli A, Miyazaki Y, Pettiti M, Matsuda M, Mahankali S, Santini E, et al. Metabolic effects of visceral fat accumulation in type 2 diabetes. J Clin Endocrinol Metab. 2002; 87(11):5098-103.
19. Marin P, Andersson B, Ottosson M, Olbe L, Chowdhury B, Kvist H, et al. The morphology and metabolism of intraabdominal adipose tissue in men. Metabolism.1992; 41(11):1242-1248.
20. Montague CT, Prins JB, Sanders L, Digby JE, O'Rahilly S. Depot‐ and sex‐specific differences in human leptin mRNA expression: implications for the control of regional fat distribution. Diabetes 1997; 46(3):342‐347.
21. Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev 2000; 21(6):697-738.
22. Bjorntorp P. Metabolic implications of body fat distribution. Diabetes Care 1991;14 (12):1132-1143.
23. Hellmer J, Marcus C, Sonnenfeld T, Arner P. Mechanisms for differences in lipolysis between human subcutaneous and omental fat cells. J Clin Endocrinol Metab. 1992; 75(1):15-20.
24. Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004; 145(5):2273‐2282.
25. Snijder MB, Dekker JM, Visser M, Bouter LM, Stehouwer CD, Kostense PJ et al. Associations of hip and thigh circumferences independent of waist circumference with the incidence of type 2 diabetes: the Hoorn Study. Am J Clin Nutr 2003; 77(5):1192-1197.
26. Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich‐Horvat P, Liu CY et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 2007; 116(1):39-48.
27. Arner P, Eckel RH. Adipose tissue as storage organ. En: George A. Bray, Claude Bouchard, WPT. James, editors. Handbook of Obesity. 1998:379-395.
28. Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009; 297(2):E271-88.
29. Bessesen DH, Robertson AD, Eckel RH. Weight reduction increases adipose but decreases cardiac LPL in reduced-obese Zucker rats. Am J Physiol Endocrinol Metab. 1991; 261(2 Pt 1): E246–E251.
30. Eckel RH, Yost TJ. Weight reduction increases adipose tissue lipoprotein lipase responsiveness in obese women. J Clin Invest.1987; 80(4):992–997.
31. Pollare T, Vessby B, Lithell H. Lipoprotein lipase activity in skeletal muscle is related to insulin sensitivity. Arterioscler Thromb 1991; 11(5):1192–1203.
32. Sadur CN, Yost TJ, Eckel RH. Insulin responsiveness of adipose tissue lipoprotein lipase is delayed but preserved in obesity. J Clin Endocrinol Metab 1984; 59(6):1176–1182.
33. Hartman AD. Lipoprotein lipase activities in adipose tissues and muscle in the obese Zucker rat. Am J Physiol Endocrinol Metab 1981; 241(2):E108–E115.
34. Terrettaz J, Cusin I, Etienne J, Jeanrenaud B. In vivo regulation of adipose tissue lipoprotein lipase in normal rats made hyperinsulinemic and in hyperinsulinemic genetically-obese (fa/fa) rats. Int J Obes Relat Metab Disord 1994; 18(1): 9–15.
35. Schwartz RS, Brunzell JD. Increase of adipose tissue lipoprotein lipase activity with weight loss. J Clin Invest 1981; 67(5):1425–1430.
36. Llorente-Cebrián S, Kulyté A, Hedén P, Näslund E, Arner P, Rydén M. Relationship between site-specific HSL phosphorylation and adipocyte lipolysis in obese women. Obes Facts. 2011; 4(5):365-71.
37. Langin D, Dicker A, Tavernier G, Hoffstedt J, Mairal A, Rydén M, Arner E, Sicard A, Jenkins CM, Viguerie N, van Harmelen V, Gross RW, Holm C, Arner P. Adipocyte lipases and defect of lipolysis in human obesity. Diabetes. 2005; 54(11):3190-7.
38. Arner P. Hunting for human obesity genes? Look in the adipose tissue! Int J Obes Relat Metab Disord. 2000; 24 Suppl 4:S57-62.
39. Valet P, Grujic D, Wade J, Ito M, Zingaretti MC, Soloveva V et al. Expression of human alpha 2-adrenergic receptors in adipose tissue of beta 3-adrenergic receptor-deficient mice promotes diet-induced obesity. J Biol Chem. 2000; 275(44):34797-802.
40. Boucher J, Castan-Laurell I, Le Lay S, Grujic D, Sibrac D, Krief S et al. Human alpha 2A-adrenergic receptor gene expressed in transgenic mouse adipose tissue under the control of its regulatory elements. J Mol Endocrinol. 2002; 29(2):251-64.
41. Frühbeck G, Gómez-Ambrosi J, Muruzábal FJ, Burrell MA. The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am J Physiol Endocrinol Metab. 2001; 280(6):E827-47.
42. Martí A, Berraondo B, Martínez JA. Leptin: physiological actions. J Physiol Biochem. 1999; 55(1):43-9.
43. Mueller WM, Gregoire FM, Stanhope KL, Mobbs CV, Mizuno TM, Warden CH et al. Evidence that glucose metabolism regulates leptin secretion from cultured rat adipocytes. Endocrinology. 1998; 139(2):551-8.
44. Wellhoener P, Fruehwald-Schultes B, Kern W, Dantz D, Kerner W, Born J et al. Glucose metabolism rather than insulin is a main determinant of leptin secretion in humans. J Clin Endocrinol Metab. 2000; 85(3):1267-71.
45. Nakata M, Yada T, Soejima N, Maruyama I. Leptin promotes aggregation of human platelets via the long form of its receptor. Diabetes. 1999; 48(2):426-429.
46. Mcgill, JB, Schneider, DJ, Arfken, CL, Lucore, CL. Factors responsable for impaired fibrinolysis in obese subject and NIDDM patiens. Diabetes 1994; 43(1): 104-109.
47. Palomer X, Pérez A, Blanco-Vaca F. Adiponectin: a new link between obesity, insulin resistance and cardiovascular disease. Med Clin (Barc) 2005; 124(10):388-95.
48. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999; 257(1):79-83.
49. Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000; 20(6):1595-9.
50. Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004; 291(14):1730-7.
51. Kazumi T, Kawaguchi A, Sakai K, Hirano T, Yoshino G. Young men with high-normal blood pressure have lower serum adiponectin, smaller LDL size, and higher elevated heart rate than those with optimal blood pressure. Diabetes Care. 2002; 25(6):971-6.
52. Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002; 277(29):25863-6.
53. Ouchi N, Kihara S, Arita Y, Nishida M, Matsuyama A, Okamoto Y et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation. 2001; 103(8):1057-63.
54. Ryo M, Nakamura T, Kihara S, Kumada M, Shibazaki S, Takahashi M et al. Adiponectin as a biomarker of the metabolic syndrome. Circ J. 2004; 68(11):975-81.
55. Kumada M, Kihara S, Sumitsuji S, Kawamoto T, Matsumoto S, Ouchi N et al. Osaka CAD Study Group. Coronary artery disease. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol. 2003; 23(1):85-9.
56. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001 Jan 18;409(6818):307-12.
57. Patel L, Buckels AC, Kinghorn IJ, Murdock PR, Holbrook JD, Plumpton C et al. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun 2003; 300(2):472-6.
58. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 1995; 95(5):2409–2415.
59. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006; 444(7121):860–867
60. Duncan ER, Walker SJ, Ezzat VA, Wheatcroft SB, Li JM, Shah AM, et al. Accelerated endothelial dysfunction in mild prediabetic insulin resistance: the early role of reactive oxygen species. Am J Physiol Endocrinol Metab. 2007; 293(5):E1311–E1319.
61. Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesitydiabetes. Central role of tumor necrosis factor. J Clin Invest 1994; 94(4):1543–1549
62. Hube F, Birgel M, Lee YM, Hauner H. Expression pattern of tumour necrosis factor receptors in subcutaneous and omental human adipose tissue: role of obesity and noninsulin-dependent diabetes mellitus. Eur J Clin Invest 1999; 29(8):672–678.
63. Ruan H, Lodish HF. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev. 2003; 14(5):447–455
64. Uysal KT, Wiesbrock SM, MarinoMW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997; 389:610–614
65. Takano M, Nishihara R, Sugano N, Matsumoto K, Yamada Y, Takane M, et al. The effect of systemic anti-tumor necrosis factor-alpha treatment on Porphyromonas gingivalis infection in type 2 diabetic mice. Arch Oral Biol. 2010; 55(5):379–384
66. Gómez-Hernández A, Otero YF, de las Heras N, Escribano O, Cachofeiro V, Lahera V, et al. Brown fat lipoatrophy and increased visceral adiposity through a concerted adipocytokines overexpression induces vascular insulin resistance and dysfunction. Endocrinology. 2012; 153(3):1242-55.
67. Van Harmelen V, Ariapart P, Hoffstedt J, Lundkvist I, Bringman S, Arner P. Increased adipose angiotensinogen gene expression in human obesity. Obes Res. 2000; 8(4):337-41.
68. Shimomura I, Funahashi T, Takahashi M, Maeda K, Kotani K, Nakamura T et al. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med. 1996; 2(7):800-3.
69. Jones BH, Standridge MK, Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology. 1997; 138(4):1512-9.
70. Gil A, María Aguilera C, Gil-Campos M, Cañete R. Altered signalling and gene expression associated with the immune system and the inflammatory response in obesity. Br J Nutr. 2007; 98 Suppl 1:S121-6.
71. Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011; 13(1):11-22.
72. Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011; 11(11):738-49.
73. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009; 15(8):914–920.
74. Crandall DL, Hausman GJ, Kral JG. A review of the microcirculation of adipose tissue: anatomic, metabolic, and angiogenic perspectives. Microcirculation. 1997; 4(2):211-32.
75. Blake GJ, Ridker PM. Inflammatory bio-markers and cardiovascular risk prediction. J Intern Med. 2002; 252(4):283-94.
76. Sengenès C, Miranville A, Lolmède K, Curat CA, Bouloumié A. The role of endothelial cells in inflamed adipose tissue. J Intern Med. 2007; 262(4):415-21
77. Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005; 11(2):183-90.
78. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006; 43(2 Suppl 1):S54-62.
79. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006; 116(7):1793-801.
80. Mattson MP. Perspective: Does brown fat protect against diseases of aging? Ageing Res Rev. 2010; 9(1):69-76.
81. Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006; 4 (4):263–73.
82. Ishibashi J, Seale P. Medicine. Beige can be slimming. Science. 2010; 328(5982):1113-4.
83. Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012; 150(2):366-76.
84. Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012; 481(7382):463-8.
85. Ghorbani M, Claus TH, Himms-Hagen J. Hypertrophy of brown adipocytes in brown and white adipose tissues and reversal of diet-induced obesity in rats treated with a beta3-adrenoceptor agonist. Biochem Pharmacol. 1997; 54(1):121-31.
86. Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest. 1998; 102(2):412-20.
87. Lowell BB, S-Susulic V, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, et al. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993; 366:740–2.
88. Guerra C, Navarro P, Valverde AM, Arribas M, Brüning J, Kozak LP, et al. Brown adipose tissue-specific insulin receptor knockout shows diabetic phenotype without insulin resistance. J Clin Invest. 2001; 108(8):1205-13.
89. Kontani Y, Wang Y, Kimura K, Inokuma KI, Saito M, Suzuki-Miura T, et al. UCP1 deficiency increases susceptibility to diet-induced obesity with age. Aging Cell. 2005; 4(3):147-55.
90. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009; 360(15):1509-17.
91. Hansen JB, Kristiansen K. Regulatory circuits controlling white versus brown adipocyte differentiation. Biochem J. 2006; 398(2):153-68
92. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004; 84(1):277-359.
93. Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011; 17(2):200-5.
94. Williams KJ. Molecular processes that handle -- and mishandle -- dietary lipids. J Clin Invest. 2008; 118(10):3247-59.
95. Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996; 3(2):213-9.
96. Austin MA, McKnight B, Edwards KL, Bradley CM, McNeely MJ, Psaty BM, et al. Cardiovascular disease mortality in familial forms of hypertriglyceridemia: A 20-year prospective study. Circulation. 2000; 101(24):2777-82.
97. Mooradian AD. Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009; 5(3):150-9.
98. Fredriksson JM, Nikami H, Nedergaard J. Cold-induced expression of the VEGF gene in brown adipose tissue is independent of thermogenic oxygen consumption. FEBS Lett. 2005; 579(25):5680-4.
99. Mitchell JR, Jacobsson A, Kirchgessner TG, Schotz MC, Cannon B, Nedergaard J. Regulation of expression of the lipoprotein lipase gene in brown adipose tissue. Am J Physiol. 1992; 263(3 Pt 1):E500-6.
100. Ouellet V, Labbé SM, Blondin DP, Phoenix S, Guérin B, Haman F, et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest. 2012; 122(2):545-52.
101. Chechi K, Blanchard PG, Mathieu P, Deshaies Y, Richard D. Brown fat like gene expression in the epicardial fat depot correlates with circulating HDL-cholesterol and triglycerides in patients with coronary artery disease. Int J Cardiol. 2012 Jun 22.
102. Shabalina IG, Jacobsson A, Cannon B, Nedergaard J. Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem. 2004; 279(37):38236-48.
103. Nikami H, Shimizu Y, Endoh D, Yano H, Saito M. Cold exposure increases glucose utilization and glucose transporter expression in brown adipose tissue. Biochem Biophys Res Commun. 1992; 185(3):1078-82
104. Dallner OS, Chernogubova E, Brolinson KA, Bengtsson T. Beta3-adrenergic receptors stimulate glucose uptake in brown adipocytes by two mechanisms independently of glucose transporter 4 translocation. Endocrinology. 2006; 147(12):5730-9.
105. Chartoumpekis DV, Habeos IG, Ziros PG, Psyrogiannis AI, Kyriazopoulou VE, Papavassiliou AG. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol Med. 2011; 17(7-8):736-40.
106. Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, et al. Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J Biol Chem. 2011; 286(15):12983-90.
107. Hondares E, Rosell M, Gonzalez FJ, Giralt M, Iglesias R, Villarroya F. Hepatic FGF21 expression is induced at birth via PPARalpha in response to milk intake and contributes to thermogenic activation of neonatal brown fat. Cell Metab. 2010; 11(3):206-12.
108. Nisoli E, Tonello C, Carruba MO. Nerve growth factor, beta3-adrenoceptor and uncoupling protein 1 expression in rat brown fat during postnatal development.Neurosci Lett. 1998; 246(1):5-8.
109. Nisoli E, Tonello C, Benarese M, Liberini P, Carruba MO. Expression of nerve growth factor in brown adipose tissue: implications for thermogenesis and obesity. Endocrinology 1996; 137(2):495-503.
110. Néchad M, Ruka E, Thibault J. Production of nerve growth factor by brown fat in culture: relation with the in vivo developmental stage of the tissue. Comp Biochem Physiol Comp Physiol. 1994; 107(2):381-8.
111. Sornelli F, Fiore M, Chaldakov GN, Aloe L. Adipose tissue-derived nerve growth factor and brain-derived neurotrophic factor: results from experimental stress and diabetes. Gen Physiol Biophys 2009; 28 Spec No:179-83.
112. Tonello C, Giordano A, Cozzi V, Cinti S, Stock MJ, Carruba MO, et al. Role of sympathetic activity in controlling the expression of vascular endothelial growth factor in brown fat cells of lean and genetically obese rats. FEBS Lett. 1999; 442(2-3):167-72.
113. Kikuchi-Utsumi K, Gao B, Ohinata H, Hashimoto M, Yamamoto N, Kuroshima A. Enhanced gene expression of endothelial nitric oxide synthase in brown adipose tissue during cold exposure. Am J Physiol Regul Integr Comp Physiol. 2002; 282(2):R623-6.
114. Fitzgibbons TP, Kogan S, Aouadi M, Hendricks GM, Straubhaar J, Czech MP. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am J Physiol Heart Circ Physiol 2011; 301(4):H1425-37.
115. Ortega MT, Xie L, Mora S, Chapes SK. Evaluation of macrophage plasticity in brown and white adipose tissue. Cell Immunol 2011; 271(1):124-33.
116. Meijer RI, Serne EH, Smulders YM, van Hinsbergh VW, Yudkin JS, Eringa EC. Perivascular adipose tissue and its role in type 2 diabetes and cardiovascular disease. Curr Diab Rep. 2011; 11(3):211-7.
117. Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009; 29(10):1458-64.
118. Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009; 119(12):1661-70.
119. Eringa EC, Bakker W, van Hinsbergh VW. Paracrine regulation of vascular tone, inflammation and insulin sensitivity by perivascular adipose tissue. Vascul Pharmacol. 2012 May-Jun; 56(5-6):204-9.
120. Rittig K, Staib K, Machann J, Böttcher M, Peter A, Schick F et al. Perivascular fatty tissue at the brachial artery is linked to insulin resistance but not to local endothelial dysfunction. Diabetologia. 2008; 51(11):2093-9.
121. Feldon SE, O'loughlin CW, Ray DM, Landskroner-Eiger S, Seweryniak KE, Phipps RP. Activated human T lymphocytes express cyclooxygenase-2 and produce proadipogenic prostaglandins that drive human orbital fibroblast differentiation to adipocytes. Am J Pathol. 2006; 169(4):1183-93.
122. Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, et al. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circ Res 2009; 104(4):541.
123. Payne GA, Borbouse L, Kumar S, Neeb Z, Alloosh M, Sturek M, et al. Epicardial perivascular adipose-derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome via a protein kinase C-beta pathway. Arterioscler Thromb Vasc Biol. 2010; 30(9):1711-7.
124. Takaoka M, Suzuki H, Shioda S, Sekikawa K, Saito Y, Nagai R, et al. Endovascular injury induces rapid phenotypic changes in perivascular adipose tissue. Arterioscler Thromb Vasc Biol. 2010; 30(8):1576–82.
125. Henrichot E, Juge-Aubry CE, Pernin A, Pache JC, Velebit V, Dayer JM, et al. Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis? Arterioscler Thromb Vasc Biol. 2005; 25(12):2594–9.
126. Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension. 2009; 54(6):1384–92.
127. Sacks HS, Fain JN. Human epicardial fat: what is new and what is missing? Clin. Exp. Pharmacol. Physiol. 2011; 38(12):879–887.
128. Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. 2003; 108(20):2460–6.
129. Redman LM, Ravussin E. Endocrine alterations in response to calorie restriction in humans. Mol Cell Endocrinol. 2009; 299(1):129-36.
130. Howell A, Chapman M, Harvie M. Energy restriction for breast cancer prevention. Recent Results Cancer Res. 2009; 181:97-111.
131. López-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006; 103(6):1768-73.
132. Liu D, Pitta M, Mattson MP. Preventing NAD(+) depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann N Y Acad Sci. 2008; 1147:275-82.
133. Kamel EG, McNeill G, Van Wijk MC. Change in intra-abdominal adipose tissue volume during weight loss in obese men and women: correlation between magnetic resonance imaging and anthropometric measurements. Int J Obes Relat Metab Disord. 2000; 24(5):607-13.
134. Seidell JC.Environmental influences on regional fat distribution. Int J Obes. 1991; 15 Suppl 2:31-5.
135. Varady KA, Bhutani S, Klempel MC, Kroeger CM. Comparison of effects of diet versus exercise weight loss regimens on LDL and HDL particle size in obese adults. Lipids Health Dis. 2011; 10:119.
136. Saris WH. Effects of energy restriction and exercise on the sympathetic nervous system. Int J Obes Relat Metab Disord. 1995; 19 Suppl 7:S17-S23.
137. Zorba E, Cengiz T, Karacabey K. Exercise training improves body composition, blood lipid profile and serum insulin levels in obese children. J Sports Med Phys Fitness. 2011; 51(4):664-9.
138. Lamarche B, Després JP, Moorjani S, Nadeau A, Lupien PJ, Tremblay A, et al. Evidence for a role of insulin in the regulation of abdominal adipose tissue lipoprotein lipase response to exercise training in obese women. Int J Obes Relat Metab Disord. 1993; 17(5):255-61.
139. Tremblay A, Nadeau A, Després JP, St-Jean L, Thériault G, Bouchard C. Long-term exercise training with constant energy intake. 2: Effect on glucose metabolism and resting energy expenditure. Int J Obes. 1990; 14(1):75.
140. Langin D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol Res. 2006; 53(6):482-91.
141. Enerbäck S. Human brown adipose tissue. Cell Metab. 2010; 11(4):248-52.
142. Nedergaard J, Bengtsson T, Cannon B. New powers of brown fat: fighting the metabolic syndrome. Cell Metab. 2011; 13(3):238-40
143. Langin D. Recruitment of brown fat and conversion of white into brown adipocytes: strategies to fight the metabolic complications of obesity? Biochim Biophys Acta. 2010; 1801(3):372-6.
144. Farmer SR. Brown fat and skeletal muscle: unlikely cousins? Cell. 2008;134(5):726-7.
145. Schulz TJ, Huang TL, Tran TT, Zhang H, Townsend KL, Shadrach JL, et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc Natl Acad Sci U S A. 2011; 108(1):143-8.
146. Townsend KL, Tseng YH. Brown adipose tissue. Recent insights into development, metabolic function and therapeutic potential. Adipocyte 2012; 1(1):13-24.