REVISIÓN |
Avances en artritis reumatoide
Luis García-Sevillano
Farmacéutico comunitario. Farmacia Navarro Gómez, Valladolid.
e-mail: sevillanolg@hotmail.com
Recibido el 16 de enero de 2014 -An. Real Acad. Farm. Vol. 80, Nº 1 (2014), pág.126-150
RESUMEN
La artritis reumatoide (AR) es la forma más frecuente de poliartritis crónica. Afecta al 0,5% de la población española. En los últimos años se han producido importantes avances en la patogénesis de la AR, en el diagnóstico y en el tratamiento de la enfermedad. Un diagnóstico temprano es esencial con el fin de prevenir la erosión articular y de mejorar la prognosis y la calidad de vida de los pacientes con AR. En esta revisión se presentan los últimos hallazgos en la epidemiología, etiología, fisiopatología, clínica, diagnóstico y tratamiento de esta enfermedad severa pero tratable. |
Palabras clave: Artritis reumatoide; Fisiopatología; Tratamiento.
aBSTRACT
Pharmacist’s update in rheumatoid arthritis
Rheumatoid arthritis (RA) is the most frequent form of chronic polyarthritis. It affects 0.5% of the Spanish population. In recent years there have been significant advances in the pathogenesis of RA, in the diagnosis and treatment of the disease. An early diagnosis is essential in order to prevent the joint erosion and improve the prognosis and quality of life of patients with RA. In this review are presented the latest findings in the epidemiology, etiology, pathophysiology, clinical features, diagnosis and treatment of this severe but treatable disease. |
Keywords: Rheumatoid Arthritis; Pathophysiology; Treatment.
INTRODUCCIÓN
La artritis reumatoide (AR) es una enfermedad compleja, de naturaleza autoinmune, sistémica, crónica, del tejido conjuntivo, que afecta sobre todo a las articulaciones periféricas y cursa con dolor e inflamación en las articulaciones, a la vez que también pueden presentarse manifestaciones extra-articulares (1-3).Tiene un gran impacto en la calidad de vida del paciente y origina un coste económico y social muy importante. En la mayoría de los pacientes el curso de la enfermedad es progresivo y conduce a una lesión estructural articular, deterioro funcional, disminución de la calidad de vida, aumento de la morbilidad y de la mortalidad. Es una enfermedad de comienzo insidioso y pueden pasar meses hasta que el paciente busque consulta médica. Es muy importante un diagnóstico y un tratamiento precoces para reducir en lo posible el daño estructural.
Epidemiología de la artritis reumatoide
La prevalencia en el mundo oscila entre el 0,5% y 1,0%. En el norte de Europa y de América (4) se estima su prevalencia en torno al 1% de la población general. Existen diferencias geográficas en cuanto a la severidad de la AR, así se ha observado que es menos severa en los países mediterráneos frente a los del norte de Europa (5). La prevalencia en España según el EPISER (6) es de 0,5%. La incidencia de la AR en España se aproxima a los 10 casos/100000 habitantes (7). La AR es más frecuente en mujeres que en hombres (2-3 mujeres por cada hombre) (8) y suele aparecer entre la cuarta y sexta década de la vida.
Etiología
La etiología de la AR es desconocida pero se postula que se desencadena en individuos con una predisposición genética después de la exposición repetida a ciertos agentes ambientales (1, 9,10).
En estudios con gemelos se ha estimado que los factores genéticos contribuyen con aproximadamente un 50% al desarrollo de la AR y el resto se atribuye a factores ambientales (11). El factor de riesgo genético (12) más importante para la aparición de la AR está asociado al complejo mayor de histocompatibilidad (CMH) y en particular al “Human Leukocyte Antigen” (HLA), al cual se le atribuye entre un 30% y un 50% del riesgo genético total (13). Los alelos HLA-DRB1 comparten un “epítopo compartido”, que se corresponde a la secuencia glutamina-leucina-arginina-alanina-alanina (QKRAA) y se asocia a un incremento de la susceptibilidad a AR y a la gravedad de la enfermedad. Recientemente se ha descubierto que de estos 5 aminoácidos, que se encuentran formando parte de la hendidura de unión de los antígenos al CMH, dos se encuentran asociados a los genes HLA-B y HLA-DP, y los tres restantes a los genes HLA-DR y estos 5 residuos explican casi completamente la asociación entre el CMH y el riesgo de presentar AR (14). Parece ser que se producirían alteraciones a nivel de la regulación post-transcripcional, dando lugar a proteínas citrulinadas, que son reconocidas como extrañas, desencadenando la producción de anticuerpos (Ac) contra estas proteínas en el inicio de la enfermedad (15). Posiblemente los linfocitos T que reconocerían estos auto-antígenos citrulinados presentados a través del CMH, estimularían a las células B en la secreción de Ac contra las proteínas citrulinadas (ACPA). Sorprendentemente se ha observado que la asociación entre los alelos HLA-DRB1 y el riesgo de presentar AR solamente tuvo lugar con la presencia de los ACPA (16,17). También se han encontrado alelos HLA-DRB1*13:01 que podrían proteger frente al desarrollo de la AR(12). Fuera de la región del CMH se han identificado otros lugares en los genes relacionados con la AR como la “peptidylargininedeiminasetype 4”(PADI4), “proteintyrosinephosphatase non-receptor type 22”(PTPN22), el antígeno 4 asociado al linfocito T citotóxico(CTLA4), “TNF Receptor Associated Factor/ Complement component 5” (TRAFI/C5), “Signal transducer and Activator of Transcription 4” (STAT4), “Tumor Necrosis Factor Alfa-Induced Protein 3” (TNFAIP3), etc. También hay una relación probable entre la AR y los genes que codifican la cascada metabólica del factor nuclear kappa B (NFKb) (CD40, CD244, CDK6, CCL21, PRKCQ, TNFRSF14, PIP4K2C, IL-1B, IL-2RB, IL-2RA) (18). Para muchos de ellos todavía no se conocen los polimorfismos funcionales (12).
En cuanto a los factores ambientales implicados en el origen de la AR, no hay consenso para ninguno de ellos (19). Algunos factores de riesgo pueden ser comunes a otras enfermedades autoinmunes. Así, en una revisión sistemática, se ha observado un incremento de la prevalencia de tiroiditis autoinmune y diabetes mellitus insulinodependiente en los pacientes con AR y una asociación inversa entre la esclerosis múltiple y AR (20). En las mujeres la AR se desarrolla frecuentemente en períodos en los cuales las hormonas esteroideas fluctúan mucho, como por ejemplo el post-parto y la perimenopausia (21). Es probable que desde antes del nacimiento empiecen a acumularse los factores de riesgo como puede ser el peso al nacer y la composición genética del CMH de la madre, que influirán en el desarrollo de la AR (22, 23). Fumar tabaco es uno de los factores ambientales más fuertemente asociados al riesgo de presentar AR. Se ha estimado que el riesgo es aproximadamente el doble en los fumadores que en los no fumadores, especialmente en los hombres que fuman bastante y con factor reumatoide (FR) positivo (24). Además aquellos fumadores y ACPA tenían una mayor susceptibilidad genética de presentar AR (genes del epítopo compartido del CMH) (25). También estudios recientes confirman una peor respuesta al tratamiento de la AR en aquellos pacientes fumadores (26). Otros factores potenciales pueden ser factores asociados al trabajo (27), como polvo de sílice, aceites minerales, etc. Algunos datos indican que un consumo moderado de alcohol puede reducir el riesgo de presentar AR (28). En cuanto a los factores dietéticos la ingesta de vitamina D está inversamente relacionada con el curso de la AR, aunque hay estudios en los que no se ha observado una relación entre los niveles en plasma de vitamina D y la presencia de los auto-anticuerpos en la AR (29, 30). Los ácidos grasos poliinsaturados de cadena larga omega 3, que se encuentran principalmente en el pescado azul, disminuyen la producción de eicosanoides, citocinas, especies reactivas de oxígeno y la expresión de moléculas de adhesión que intervienen en la inflamación (31). Los ácidos grasos omega 3 intervienen en la producción de las resolvinas (una familia de mediadores antiinflamatorios). Estudios de caso-control y de cohortes han mostrado un efecto protector modesto de los ácidos grasos omega 3 marinos en el riesgo de desarrollar AR (32). Además, el consumo de pescado también ha demostrado mejorar los síntomas de la AR y su progresión (33). Parece que los antioxidantes podrían proteger frente a la aparición de la AR, pero los resultados para los antioxidantes por separado son contradictorios (34). Otros posibles factores de riesgo que se han investigado bastante en su asociación con la AR son las infecciones víricas (virus de Epstein-Barr, parvovirus humanos B-19) y bacterianas (Proteusmirabilis, micobacterias, micoplasmas, etc.). Recientemente se ha investigado el papel de una bacteria causante de infecciones periodontales, Porphyromonas gingivalis, la cual parece ser bastante prevalente en los estadios iniciales de la AR (35). P. gingivalis es la única bacteria que causa infecciones en humanos y que contiene la enzima peptidil arginina deiminasa, la cual tiene la capacidad de citrulinar las proteínas (36). Estas proteínas citrulinadas podrían favorecer la producción de ACPA y desencadenar la AR, aunque se necesitan más investigaciones al respecto (37). También se ha estudiado la relación entre la obesidad y la AR por varios investigadores. Se ha observado un incremento de la morbilidad y mortalidad cardiovascular en aquellos pacientes con AR (38). Ajeganova et al. (39) y Wolf y Michaud (40) mostraron que la obesidad está asociada a un empeoramiento de la clínica de la AR. Además, Stavropoulos-Kalinoglou et al. (41) pusieron de manifiesto que mientras la terapia anti-TNF (Factor de Necrosis Tumoral) mejoraba la sensibilidad a la insulina en los pacientes con AR y normopeso, no ocurría lo mismo en los pacientes con sobrepeso.
Fisiopatología de la AR
La AR se caracteriza por la inflamación de la membrana sinovial, seguida de la formación de un tejido que se llama “pannus” y que conduce a la destrucción del cartílago y erosión de los huesos con la consiguiente limitación funcional, junto con las manifestaciones extraarticulares.
Parece ser que una producción de auto-Ac (ACPA, FR) e inflamación sistémica precederían a la inflamación sinovial; en algunos pacientes hasta 15 años antes (42).
En la inflamación de las articulaciones intervienen una serie de cascadas inflamatorias, desencadenadas probablemente por la inmunidad adaptativa, tanto en los pacientes que presentan o no el FR o ACPA. En la AR se ha observado que las células progenitoras hematopoyéticas presentan un acortamiento telomérico inapropiado para su edad que podría producir envejecimiento prematuro y una posible restricción de la capacidad proliferativa (43). Este defecto parece estar específicamente conectado con la cascada de señalización ERK (44).
La membrana sinovial es el lugar donde se inicia el proceso inflamatorio articular y está compuesta por una población de células entre las que destacan los fibroblastos y los macrófagos. La inflamación de la membrana sinovial se produce tras la infiltración en la misma de linfocitos T (45) y B (46), células endoteliales y macrófagos activados, que van a producir una serie de mediadores solubles, en su mayoría citocinas (47, 48), pero también factores de crecimiento, quimiocinas y moléculas que participan en la señalización intracelular (Figura 1). Las vías de transducción de señales intracelulares también pueden estar implicadas en la patogénesis de la AR (vías de la quinasa Janus, quinasas proteínicas activadoras de mitógenos, NFKb, quinasa c-Jun N-terminal, RANKL, ligando del receptor activador del factor nuclear kappa B, etc.).
Las citocinas son proteínas o glucoproteínas de bajo peso molecular que tienen una vida media corta, que son producidas principalmente por las células del sistema inmunológico y que actúan como mediadores fundamentales en la transmisión de señales intercelulares.
Dentro de los linfocitos T, la infiltración en particular de los linfocitos Th 17 que son secretores de la citocina con mayor efecto proinflamatorio, la IL-17; parece ser la responsable de la iniciación del proceso inflamatorio al interaccionar con CD, macrófagos y linfocitos B (Figura 1). La IL-17 induce la degradación directa de los proteoglucanos del cartílago. La IL-17 induce la expresión del ligando RANKL en los fibroblastos sinoviales, que van a provocar la diferenciación de los osteoclastos, que migran hacia la zona marginal iniciando la erosión ósea (49, 50). La IL-17 desencadena un incremento en la inflamación local a través de citocinas inflamatorias como el TNFa, IL-1 e IL-6.
Las células Th1 y Th2 tienen efectos inhibitorios en la osteoclastogénesis a través de la secreción de interferon-γ (IFN-γ) e IL-4 respectivamente (Figura 2).
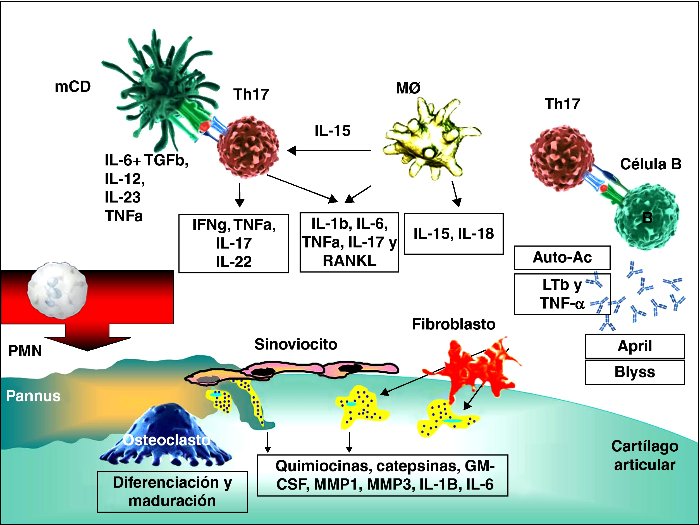
Figura 1.- Esquema de la fisiopatología de la AR. Interacción de las principales células implicadas en la fisiopatología de la sinovitis y resorción ósea, culminando en la liberación de enzimas y daño tisular (CD: células dendríticas, MF: macrófagos, GM-CSF: factor estimulador de la colonias de macrófagos y granulocitos, MMP: metaloproteinasa, IL: interleucina, LTb: linfotoxina beta, TGFb: Transforming Growth Factor beta, April: ligando inductor de la proliferación del linfocito B, Blyss: factor estimulador del linfocito B) (48).
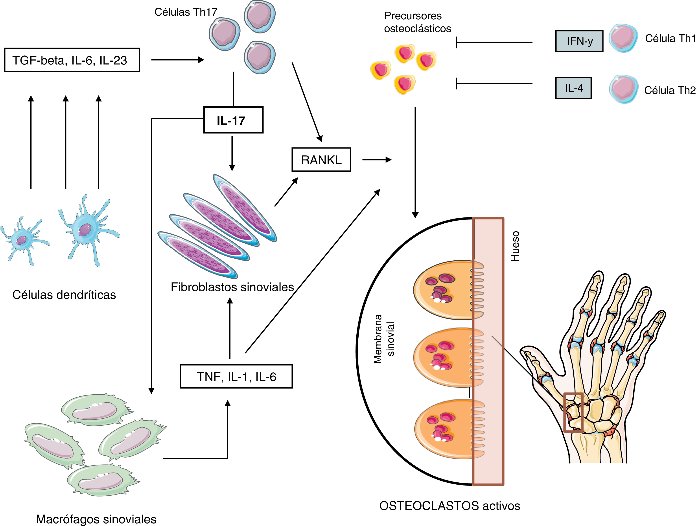
Figura 2.- Patogenia de la erosión reumatoide (IFN-g: interferón gamma) (50).
Los macrófagos son células fundamentales implicadas en la fisiopatología, probablemente debido a la secreción de mediadores proinflamatorios claves como el TNF-a y la IL-1, que perpetúan de forma crónica la inflamación en la AR (51). Los fibroblastos sinoviales son activados por este microambiente local transformándose y dando lugar a un fenotipo pseudomaligno con regulación al alta de oncogenes, inhibición de la apoptosis y secreción de citocinas, quimiocinas, metaloproteinasas de la matriz y catepsinas, que median el proceso inflamatorio crónico y la destrucción articular (52) (Figura 2). Los linfocitos B actúan mediante la producción de auto-Ac (FR, ACPA); como células activadoras de los linfocitos T actuando como células presentadoras de antígeno (APC) y de activación de los fibroblastos a través de la secreción de TNF-a y linfotoxinab. La activación de los linfocitos T sólo tiene lugar en presencia de señales co-estimuladoras mediadas por la familia de receptores CD28-B7 (CD80/86). Los neutrófilos presentes en el fluido sinovial, sintetizan prostaglandinas inflamatorias, proteasas y especies reactivas de oxígeno. Además, se produce una hiperplasia y activación de los mastocitos a nivel articular. Los mastocitos son las células que presentan más receptores para la IL-17a en la membrana sinovial (53). La función de las células T CD4+ reguladoras está disminuida, lo que contribuye al desequilibrio entre los brazos efector y regulador de la inmunidad.
La angiogénesis (formación de nuevos capilares o neovascularización) es un proceso precoz y crítico en la fisiopatología de la AR. La IL-17 es capaz de incrementar la vascularización promoviendo directamente el crecimiento de los vasos sanguíneos, estimulando los fibroblastos sinoviales que van a secretar el factor de crecimiento endotelial vascular y activando las células endoteliales que migrarán por quimiotaxis y darán por resultado la neovascularización.
El TNF-a y la IL-6 son citocinas proinflamatorias que actúan sinérgicamente como moléculas efectoras fundamentales y son los principales componentes del proceso inflamatorio. El TNF-a es un mediador importante para la producción de citocinas, metaloproteinasas, óxido nítrico, prostaglandina E2, etc. La IL-6 es una citocina proinflamatoria pleiotrópica con efectos variados sobre las líneas hematopoyéticas, como la inducción de la producción de Ig por las células B, proliferación de las células T, inducción de la síntesis de las proteínas de fase aguda hepáticas y diferenciación en las células T citotóxicas.
La IL-1b participa en la destrucción del cartílago y hueso a través de la secreción de metaloproteinasas, disminución de la síntesis de glucosa minoglicanos, etc. (54). Los inhibidores fisiológicos de las citocinas proinflamatorias tales como el antagonista del receptor de la IL-1(IL1-Ra), sIL1-RI, sIL1-RII, sTNFRI, sTNF-RII se encuentran en mayor cantidad pero no la suficiente como para contrarrestar la inflamación.
La IL-23 es uno de los factores esenciales para la supervivencia y/o expansión de los linfocitos Th17 y, por otra parte, induce la secreción de IL-17 por células no T. La interacción entre la IL-23 y la IL-17 desempeña un papel fundamental tanto en las fases precoces de la AR como en la fase destructiva, ya que induce la expresión del receptor activador del factor nuclear kappa B (RANK) en precursores mieloides y de su ligando RANKL en linfocitos colaboradores (Th). Además, la IL-23 estimula la producción de IL-1b y TNF-a.
La IL-18 induce la activación de los linfocitos T citotóxicos y células Natural Killers (NK) (55), induciendo a su vez la secreción de INF-g por dichas células.
La IL-27 se produce en las APC en presencia de infección e INF-a. Induce efectos pleiotrópicos sobre diversas células del sistema inmunológico y tiene propiedades pro- y anti-inflamatorias (56).
La IL-32 es una citocina proinflamatoria que se ha encontrado en células NK y en linfocitos T activados.
La IL-35 es una citocina perteneciente a la familia de la IL-12; se produce en los linfocitos T CD4+ reguladores y se ha visto que in vitro es capaz de inhibir la actividad de los linfocitos Th 17.
Otras citocinas importantes en la patogenia de la AR son el factor estimulador del linfocito B (BLyS) y el ligando inductor de la proliferación del linfocito B (APRIL). El bloqueo de estas citocinas podría ser importante en el tratamiento de la AR.
PRESENTACIÓN CLÍNICA
La presentación clínica de la AR es extremadamente variable, mostrando un amplio espectro de manifestaciones clínicas variando desde manifestaciones de la enfermedad moderadas a rápidamente presentar inflamación progresiva, destrucción articular y una discapacidad física severa.
La enfermedad comienza con dolor y tumefacción articular que se puede instaurar en semanas o meses. Las articulaciones más afectadas al inicio de la enfermedad son las metacarpofalángicas (MCF), interfalángicas proximales (IFP) de las manos, muñecas y metatarsofalángicas (MTF) de los pies. A medida que avanza la AR se afectan otras articulaciones como hombros, codos, rodillas y tobillos. La rigidez matutina es característica de la AR activa y se define como la dificultad al movimiento de las articulaciones al levantarse de la cama o después de permanecer un tiempo en reposo. La afectación articular es simétrica y la rigidez matutina de más de 1 hora de duración. Además de estos síntomas articulares el paciente puede tener síntomas generales como fiebre, fatiga, pérdida de peso, mialgias y afectación emocional (57). La fatiga es un estado fisiológico causado por la acción directa de citocinas proinflamatorias (en particular la IL-1 e IL-6) sobre receptores de citocinas en las células endoteliales del cerebro (58). Por tanto, la fatiga debe ser medida como una parte del estado del paciente, y así se ha observado un efecto beneficioso disminuyendo la fatiga en los nuevos tratamientos con antagonistas de citocinas (59). La afectación extraarticular puede afectar al 50% de los pacientes. La manifestación más frecuente es el síndrome de Sjögren (35%), que se caracteriza por sequedad bucal y ocular. Los nódulos reumatoides se presentan sobre superficies de presión como codos, tendón de Aquiles y dedos. El 7% de los pacientes en el momento del diagnóstico de la AR tienen nódulos y aproximadamente un 30% de los pacientes tendrán nódulos en algún momento de la enfermedad. Los nódulos reumatoides aparecen más frecuentemente en aquellos pacientes que tienen el FR. Las manifestaciones extra-articulares más severas como vasculitis, enfermedad pulmonar intersticial, pericarditis y pleuritis son más frecuentes en aquellos pacientes con nódulos reumatoides. Otras manifestaciones son neuropatía asociada a vasculitis, alteraciones de la fórmula sanguínea y de las transaminasas, atrofia muscular, síndrome de Felty, glomerulonefritis y escleritis (3).
DIAGNÓSTICO
Entre los factores predictivos para el diagnóstico de la AR desde una artritis inflamatoria periférica tenemos la edad avanzada, sexo femenino y rigidez articular. La artritis inflamatoria evoluciona a AR cuando se ven afectadas un número elevado de articulaciones dolorosas y tumefactas, tanto grandes como pequeñas articulaciones, tanto de extremidades superiores como inferiores y simétricamente (60). No hay ningún test específico para el diagnóstico de la AR. El diagnóstico se lleva a cabo en base del fenotipo clínico (poliartritis simétrica crónica) y con la ayuda de datos analíticos o radiográficos (erosión articular). Los criterios de clasificación diagnóstica para la AR se desarrollaron en 1987 (61) por el Colegio Americano de Reumatología (ACR) y hasta ahora se han usado principalmente para estudios epidemiológicos o ensayos clínicos más que para la práctica clínica. Estos criterios surgieron a partir de pacientes con una AR establecida y tenían una muy pobre sensibilidad para el diagnóstico de AR en pacientes con comienzo reciente de la artritis (62).
Un diagnóstico y un tratamiento precoz efectivo en las primeras fases de la enfermedad reducen el daño estructural (63,64). La mayoría de los pacientes tienen un daño radiológico en los primeros 2 años de la enfermedad y es en este período cuando el daño estructural avanza con más rapidez. Toda artritis de más de 4 semanas de duración debe ser referida a atención especializada y en caso de sospecha de una artritis séptica la derivación será inmediata.
En 2010 se han establecido nuevos criterios de clasificación para el diagnóstico de la AR gracias al consenso entre la ACR y la liga europea contra el reumatismo (EULAR) (65). Algunos ítems incluidos en los criterios ACR de 1987, como por ejemplo los nódulos reumatoides, la presencia de erosiones radiográficas y la presencia de artritis simétrica, no se han incluido dentro de las últimas recomendaciones en 2010; sin embargo otros, como la presencia de ACPA, han sido incorporados. Estos nuevos criterios tienen una mayor sensibilidad en el diagnóstico de la AR de reciente comienzo pero una menor especificidad y por tanto una mayor cantidad de falsos positivos (66, 67). Por lo tanto se debe llevar a cabo un diagnóstico diferencial entre la AR de reciente comienzo y otras variantes de la artritis como el lupus eritematoso sistémico, artritis psoriásica, artritis microcristalina y osteoartritis entre otras. Sin embargo, más de la mitad de los pacientes con artritis temprana no desarrollarán la AR y en algunos casos de artritis no clasificable podría remitir espontáneamente (68). La investigación se ha centrado en identificar aquellos factores en pacientes con artritis inflamatoria de comienzo reciente, que puedan predecir la instauración de una artritis persistente o una verdadera AR. Alguno de estos factores ha sido identificado, siendo la presencia de ACPA uno de los más importantes (69). Recientemente se ha descubierto que el genotipo TT del polimorfismo STAT4 rs7574865 está asociado a una actividad alta de la enfermedad y discapacidad en pacientes con artritis temprana (70).
Criterios para la clasificación de Artritis Reumatoide 2010 ACR/EULAR
La población objetivo está formada por aquellos pacientes que tienen al menos una articulación con sinovitis clínica definitiva (edema) y aquellos pacientes con sinovitis no explicada mejor por otra enfermedad. Para clasificar a un paciente con AR definitiva se necesita una puntuación ≥6/10 (suma de las categorías A-D, ver Tabla 1). Aquellos pacientes con una puntuación <6/10 no pueden clasificarse como AR, su condición debe ser reevaluada y los criterios pueden cumplirse acumulativamente con el tiempo.
Tabla 1. -Criterios para la clasificación de Artritis Reumatoide 2010 ACR/EULAR.
A. Compromiso articular |
|
1 articulación grande |
0 |
2-10 articulaciones grandes |
1 |
1-3 articulaciones pequeñas (con o sin compromiso de articulaciones grandes) |
2 |
4-10 articulaciones pequeñas (con o sin compromiso de grandes articulaciones) |
3 |
>10 articulaciones (al menos 1 articulación pequeña) |
5 |
B. Serología (al menos 1 resultado de la prueba es necesario para la clasificación) |
|
FR negativo y anti-CCP negativo |
0 |
FR débil positivo o anti-CCP débil positivo |
2 |
FR fuerte positivo o anti-CCP fuerte positivo |
3 |
C. Reactantes de fase aguda (al menos 1 prueba es necesaria para la clasificación) |
|
PCR normal y VSG normal |
0 |
PCR elevada o VSG elevada |
1 |
D. Duración de los síntomas |
|
<6 semanas |
0 |
≥6 semanas |
1 |
Análisis de laboratorio.
Las enfermedades autoinmunes como la AR se suelen caracterizar por la presencia de auto-Ac. El FR es un anticuerpo IgM dirigido contra la IgG. El FR no es específico para la AR y puede estar presente en pacientes con otras enfermedades, tales como infecciones (hepatitis C), procesos inflamatorios y en personas mayores saludables. Se detecta en el 75-85% de pacientes durante el curso de la enfermedad. Su sensibilidad oscila entre el 40-80%. El ACPA es más específico para la AR (63-100%) y podría jugar un papel importante en la patología de la enfermedad (71). Aproximadamente del 50 al 80 por ciento de las personas con AR tienen FR, ACPA o ambos (2). La positividad del ACPA en AR temprana incrementa el riesgo de progresión del daño articular y pueden predecir mejor que el FR una enfermedad erosiva. Recientemente, aparte de los factores de riesgo genéticos, como los alelos del epítopo compartido HLA-DRB1, PTPN22 y fumar en el desarrollo de la AR, se ha observado la presencia de ACPA alfa-enolasa y vimentina (72).Los pacientes con AR podrían tener un anticuerpo antinuclear, sobre todo en las formas de AR juvenil (73).
Los reactantes de fase aguda: el volumen de sedimentación globular (VSG) y la proteína C Reactiva (PCR) reflejan la actividad inflamatoria, tanto la presencia como la intensidad de ésta, pero no tienen valor diagnóstico. Se ha observado una relación entre los niveles elevados de reactantes de fase aguda especialmente la PCR, de forma mantenida y peor pronóstico de la enfermedad (74).
Recientemente se han descubierto Ac contra las proteínas carbamiladas en pacientes con AR. Estos Ac también están presentes en alrededor del 20% de aquellos pacientes con AR y negatividad frente a ACPA (75).
INDICES DE ACTIVIDAD DE LA ENFERMEDAD
Tanto la evaluación inicial como las de seguimiento de la AR se apoyarán en la valoración sistemática de un conjunto mínimo de parámetros que permitan evaluar el grado de actividad inflamatoria, de discapacidad funcional y de daño estructural. Este conjunto mínimo de parámetros para evaluar la AR recomendada por OMERACT (76) (outcome measures in rheumatoid arthritis clinical trials) son:
Número de articulaciones dolorosas.
Número de articulaciones tumefactas.
Dolor (mediante escala horizontal analógica visual de 10 cm).
Evaluación global de la enfermedad por el paciente y por el médico (escala analógica horizontal visual de 10 cm con indicador en los extremos que marquen “0” muy bien y “10” muy mal).
Reactantes de fase aguda que incluyen la VSG y PCR.
Capacidad funcional física mediante cuestionarios validados como el HAQ (Health Assessment Questionnaire) (77).
Daño radiológico.
La utilización de índices de actividad que resuman la
información de varios parámetros en un solo indicador es un procedimiento útil
y válido en la evaluación de la actividad de la enfermedad (78). El DAS
(Rheumatoid Arthritis Disease Activity Score) es uno de ellos e inicialmente
incluía 44 articulaciones. El DAS28 (79) es un DAS modificado basado en el
recuento de 28 articulaciones dolorosas y tumefactas (NAD28/NAT28). El DAS28 es
mucho más útil en la práctica clínica e incluye además la evaluación global de
la enfermedad por el paciente (EGP) y la VSG. Se calcula mediante una fórmula
matemática:
DAS28=0,56(√NAD28)+0,28(√NAT28)+0,70(ln VSG)+0,014(EGP).
En fechas recientes se ha propuesto otro índice similar, el SDAI (Simplified Disease Activity Index). Se calcula mediante una simple suma aritmética del número de articulaciones dolorosas y tumefactas, con índices reducidos de 28 articulaciones, valoración de la actividad por el paciente y el médico (medidos de 0 a 10) y la concentración de PCR en mg/L.SDAI= NAD28+NAT28+EGP+EGM+PCR (mg/dl).
CDAI (Clinical Disease Activity Index) es igual que el SDAI con la diferencia de que no se incluye la PCR.
Criterios de ACR para medir la actividad de la enfermedad (80). Se basan en 7 puntos: número de articulaciones inflamadas/tumefactas, actividad de la enfermedad evaluada por el médico y por el paciente, valoración del dolor/función física por parte del paciente y niveles de los reactantes de fase aguda. El ACR 20 supone una mejora igual o mayor al 20% en los anteriores criterios en la actividad de la enfermedad.
TRATAMIENTO
El objetivo terapéutico según la Sociedad Española de Reumatología (SER) es conseguir la remisión de la enfermedad o, en su defecto, disminuir la actividad inflamatoria de tal manera que el paciente experimente una mejoría significativa de sus síntomas, su capacidad funcional y laboral y su calidad de vida y que al mismo tiempo se retrase la lesión estructural articular.
Aunque no hay cura para la AR en el momento actual, varios estudios recientes confirman que un número significativo de pacientes podrían lograr la remisión clínica (ausencia de dolor e inflamación en las articulaciones) después de una terapia antirreumática efectiva. En algunos estudios, la tasa de remisión es alta (50-65%) (81), aunque la tasa oscila entre el 20 y el 35% en la mayoría de los estudios observacionales (82); en el caso de la remisión permanente las cifras son más bajas (83). La definición de remisión es controvertida, ya que las tasas de remisión van a depender del método empleado para estudiar la remisión (84). Sin embargo, se conoce que una significante proporción de pacientes en remisión clínica tienen sinovitis subclínica confirmada por ultrasonidos; lo cual explicaría por qué en un 10-20% de los pacientes en remisión clínica hay una progresión en el daño articular radiológico (85).
En España, aunque hay una guía de práctica clínica en AR, se ha observado que existe variabilidad en cuanto a su manejo (86).
Tratamiento no farmacológico
La educación de los pacientes acerca de las características fisiopatológicas de la enfermedad, habilidades para manejarse en el día a día y la protección de las articulaciones conducen a una mejoría en la salud de los pacientes (87). También la terapia ocupacional es beneficiosa, ya que se instruye al paciente acerca de cómo proteger las articulaciones y en el uso de material ortopédico para aumentar su calidad de vida (88). La terapia cognitiva-conductual también puede beneficiar a los pacientes con fatiga, enseñándoles cómo manejarse por sí mismos y reduciendo su dependencia (89). El paciente debe ser tratado por un equipo multidisciplinar en el que intervengan reumatólogos, enfermeras, farmacéuticos, psicólogos, etc.
Estas medidas se pueden recomendar a cualquier paciente con AR controlada por el reumatólogo.
-Descanso nocturno de diez horas como mínimo. Durante el día también es necesario descansar y hacer una hora de siesta.
-Dormir en una cama dura para evitar posibles deformaciones.
-Dormir sin almohada o con una delgada y dura, ya que así se consigue mantener todos los segmentos corporales extendidos.
-Cuando haya inflamación articular se puede recurrir a las férulas de reposo para garantizar la inmovilidad articular.
-Mover las articulaciones afectadas para mantener la flexibilidad. Realizar ejercicio físico de forma regular, adaptado a las necesidades de cada caso, sin forzar las articulaciones (natación, bicicleta, etc.) es muy saludable (90).
-Ducharse con agua caliente después de levantarse porque así se relaja la musculatura, y se consigue disminuir el periodo de rigidez matutina y favorece el movimiento.
-Dieta mediterránea y saludable, tomando al menos 3 raciones/semana de pescado azul (91).
-Suplementación con probióticos sobre todo tras tratamientos antibióticos prolongados, infecciones por hongos, gastroenteritis, etc. Se está investigando cómo el microbioma podría desempeñar un papel importante en la patogénesis de las enfermedades reumáticas (92).
Tratamiento farmacológico
El tratamiento de la AR incluye:
-Analgésicos/AINES. Actúan inhibiendo la ciclooxigenasa, reducen el dolor y la inflamación. Se deben utilizar a las dosis más bajas posiblesy durante el menor tiempo posible de acuerdo a la situación del paciente. Se asociará gastroprotección si es necesaria en caso de presentar factores de riesgo y se prescribirán teniendo en cuenta su perfil de toxicidad gástrica y cardiovascular.
-Corticoides (93). Tienen propiedades antiinflamatorias e inmunosupresoras que resultan de la inhibición de cascadas de mediadores inmunológicos como el NF-κB. Se deben utilizar dosis bajas asociadas a fármacos antirreumáticos modificadores de la enfermedad (FAME), en periodos cortos (<10 mg de prednisona o equivalente) para aliviar los síntomas y más a largo plazo para minimizar el daño radiológico. En cuanto a la vía intraarticular produce alivio rápido en articulaciones centinela. No se recomienda más de 3-4 infiltraciones al año en la misma articulación. Sin embargo sólo se recomienda en aquellos pacientes con factores de riesgo quirúrgicos significativos o con leves lesiones de la articulación tras fallo de la terapia conservadora (94).
-FAME: son los que en estudios controlados han demostrado que enlentecen o detienen la progresión de la enfermedad. El metotrexato (MTX) y leflunomida (LEF) destacan por su eficacia, rapidez de acción, influencia en la evolución de las lesiones radiográficas y tolerabilidad. Los efectos clínicos del MTX pueden atribuirse a que actúa promoviendo la apoptosis de linfocitos activados (aquellos que median los efectos a través de especies reactivas de oxígeno y dependientes de la quinasac-JunN-terminal); a un efecto antiinflamatorio dependiente de la adenosina (p. ej. reducción de la activación de los neutrófilos, disminución de la permeabilidad vascular y aumento de la función barrera endotelial); a la modulación de citocinas debido a la inhibición competitiva de la dihidrofolatoreductasa (reducción de los niveles de IL-4, IL-6, IL-13, TNF-a, IFN-g y del GM-CSF). Se emplea a dosis bajas a las que los efectos secundarios como citotoxicidad y citopenia son mínimos. Además, se recomienda utilizar ácido fólico (5 mg/semanales) para contrarrestar los efectos adversos del MTX (95). El tratamiento inicial debe incluir uno de los FAME relevantes como el MTX en escalada rápida: 7,5 mg semanales durante 1 mes y si al mes persiste la artritis se aumenta a 15 mg y si al mes todavía persiste se aumenta a 20 mg. En caso de respuesta insatisfactoria a MTX (96), alcanzadas las dosis máximas y asegurada la biodisponibilidad del agente, se recomienda utilizar LEF ó Sulfasalazina (SSZ) o un agente anti-TNF como segundo escalón terapéutico, en terapia de sustitución o en adición al MTX. En caso de toxicidad relevante al MTX que obligue a su suspensión, se recomienda utilizar LEF o SSZ o un agente anti-TNF como segundo escalón terapéutico.
No se excluye la utilización de otros FAME (antipalúdicos, ciclosporina o azatioprina), pero no se considera indispensable su uso antes de iniciar un tratamiento biológico. Todos los FAME han demostrado ser más eficaces que el placebo. Sin embargo, no existen ensayos clínicos que hayan comparado todas las combinaciones posibles de fármacos en monoterapia o terapia combinada. No hay evidencia de que una combinación sea superior a otra (97).
Terapia biológica
Se considera candidato a terapia biológica todo paciente que no haya conseguido el objetivo terapéutico con al menos un FAME relevante, preferiblemente MTX o LEF, en monoterapia o combinación y a dosis óptimas. Esto se debe a alguno de los inconvenientes de la terapia biológica como son la administración intravenosa con alguno de ellos, su elevado coste y a los efectos adversos asociados a estas terapias (98).
Según el consenso de la SER (2010) sobre la terapia biológica, el tratamiento más aconsejable es un FAME (MTX) en combinación con fármacos biológicos como los anti-TNF y tocilizumab desde el momento más precoz posible.
Las dianas terapéuticas más comunes son citocinas, células B y moléculas co-estimuladoras. Dentro de las anti-citocinas tenemos el TNF-α, IL-1, IL-6. En el tratamiento contra las células B tenemos Ac anti-CD20 y moduladores de los receptores de las células B como los estimuladores de los linfocitos B.
El tratamiento precoz con estos fármacos es capaz de inducir remisión en una proporción considerable de pacientes (50-60%), evitar el desarrollo de lesiones radiográficas o detener su progresión. Por ello se pueden usar de inicio en combinación con MTX o en monoterapia en pacientes con AR de menos de 1 año de evolución si se sospecha una evolución especialmente grave. Sin embargo hay que tener en cuenta que no se consigue una respuesta óptima en alrededor de un 40-50% de los pacientes, además muchos de estos fármacos dejan de ser eficaces con el tiempo. Algunos fármacos biológicos son específicos del tratamiento de la AR y otros no. En la actualidad existen 9 agentes biológicos comercializados en España, 5 anti-TNF y otros 4 dirigidos a otras dianas terapéuticas (99).
Anti-TNF:
Infliximab: Ac monoclonal IgG1 humano-murino quimérico recombinante frente al TNF-α. Se administra por vía intravenosa (i.v.) a dosis de 3 mg/kg cada 8 semanas. Utilizado concomitante con el MTX produce mejoría clínica en pacientes con enfermedad activa resistente a fármacos convencionales y además se reduce la inmunogenicidad del Infliximab (100).
Etanercept: proteína de fusión humana recombinante compuesta por el receptor p75 del factor de necrosis tumoral y la porción Fc de la IgG1 humana. Bloquea al TNF-α a través de una unión competitiva a su receptor. Se administra de forma subcutánea 40 mg 2 veces por semana demostrando efecto positivo en AR activa.
Adalimumab: Ac monoclonal humano recombinante (IgG1) que se administra en inyección subcutánea quincenal (40 mg). Adalimumab bloquea el TNF-α a través de la interacción con las moléculas p55 y p75 de la superficie celular de los receptores 1 y 2 del TNF. También en presencia del complemento puede provocar la lisis de las células que expresan el TNF. Además el adalimumab disminuye la producción de otras citocinas proinflamatorias como la IL-6, IL-8 y el GM-CSF. En combinación con metotrexato, está indicado para el tratamiento de la AR activa de moderada a grave en adultos, cuando la respuesta a un FAME incluido MTX haya sido insuficiente. También está indicado para el tratamiento de la AR activa, grave y progresiva en adultos no tratados previamente con MTX.
Golimumab: Ac monoclonal IgG1 humano recombinante anti-TNF-α que actúa uniéndose a las moléculas p55 y p75 de la proteína de fusión del receptor del TNF-α y neutraliza la expresión, inducida por el TNF-α en la superficie de las células endoteliales humanas, de la molécula de adhesión selectina-E, de la molécula de adhesión de células vasculares (VCAM-1) y de la molécula de adhesión intercelular (ICAM-1). Consigue mejoría sostenida de los signos y síntomas de los pacientes con AR activa de moderada a severa. Ha demostrado eficacia en pacientes con fracaso al MTX o que no habían usado MTX y en pacientes que previamente habían utilizado otro agente anti-TNF. Administración subcutánea una vez al mes.
Certolizumab: inhibidor selectivo del TNF-α, formado por un fragmento de un Ac humanizado recombinante conjugado con polietilenglicol (pegilación). La pegilación aumenta el tiempo de vida media de la molécula al reducir su aclaramiento renal, reduce la proteólisis y la inmunogenicidad. No contiene fracción Fc, por lo tanto no activa el complemento ni inicia la citotoxicidad dependiente del complemento. Es preferible para usar en el embarazo ya que no atraviesa la placenta. Se administra de forma subcutánea 200 mg cada 2 semanas. Reduce la tasa de progresión del daño articular y mejora la función física cuando se administra en combinación con MTX.
Tocilizumab(101): Ac monoclonal IgG1 humano recombinante frente al receptor de la IL-6. Inhibe la cascada de señalización de la IL-6 (102) y neutraliza totalmente las acciones derivadas de la presencia de la IL-6. Está indicado en AR activa de moderada a grave en combinación con MTX (salvo contraindicación) tras respuesta inadecuada o intolerancia a FAME/s o antagonista del TNF-a . Se administra por vía i.v. a dosis de 8 mg/Kg cada 4 semanas.
Rituximab: Ac monoclonal IgG1 humano-murino quimérico recombinante anti CD-20. CD-20 es un Ag específico que se expresa en la superficie de las células B. Rituximab produce depleción de linfocitos B sin afectar a los precursores ni a células plasmáticas. Está indicado en AR activa grave en combinación con MTX (salvo contraindicación) tras una respuesta inadecuada o intolerancia a FAME/ s, incluyendo uno o más antagonistas del TNF. Se administra en ciclos de tratamiento de 2 infusiones por vía i.v. de 1.000 mg separados 2 semanas.
Abatacept: proteína de fusión totalmente humana formada por el dominio extracelular del CTLA- 4 y el fragmento Fc de la IgG1 humana. Las células T requieren dos señales distintas para una activación completa. La primera señal es la interacción entre el péptido antigénico presentado por el CMH en la superficie de las APC y el receptor de la célula T. La segunda señal deriva de la unión entre un ligando en la APC al receptor co-estimulador en la célula T; la interacción entre de la CD28 en las células T con la CD80 o CD86 en la APC es un ejemplo clave de una señal co-estimuladora. Abatacept inhibe la unión del CD28 con el CD80 bloqueando la señal de coestimulación de los linfocitos T. Es el primer biológico que actúa mediante la interrupción de la activación de los linfocitos T. En pacientes con respuesta insuficiente a los anti-TNF, abatacept (103) combinado con MTX ha demostrado ser clínicamente superior al MTX en monoterapia. Se administra vía i.v. cada 4 semanas.
Anakinra: es una forma recombinante no glicosilada, antagonista del receptor humano de la IL-1 (IL-1Ra). Se diferencia de la forma nativa humana en que se ha añadido un residuo de metionina en el extremo amino-terminal. Es el primer agente biológico para modificar la respuesta inmunitaria biológica de la IL-1. Anakinra bloquea la actividad de la IL-1a y de la IL-1b por una unión competitiva al receptor IL-1R. La IL-1 al unirse a su receptor desencadena una respuesta inflamatoria y estimula la formación de los osteoclastos que van a exacerbar la inflamación. En varios estudios se ha visto que mejora los signos y síntomas de la AR así como la progresión radiográfica. Aunque no se ha comparado con otros biológicos, hay una percepción de que su eficacia es inferior a la de los anti-TNF. Se administra por vía subcutánea una vez al día.
Otros fármacos biológicos en investigación o todavía no aprobados en Europa
Tofacitinib (104) (Xeljanz®) es el primer medicamento aprobado por la FDA como inhibidor de la quinasa Janus (JAK). Este medicamento está indicado vía oral para el tratamiento de AR activa que no responde adecuadamente al MTX y/o otros FAME incluidos los anti-TNF.
Belimumab (105) es un Ac monoclonal totalmente humanizado que inhibe el factor estimulador de los linfocitos B (BlyS) y que está siendo desarrollado para el tratamiento de la AR. El BlyS controla el desarrollo y supervivencia de las células B. En la AR parece ser que están elevados los niveles de BlyS.
Ocrelizumab es un Ac monoclonal IgG1 humanizado frente al Ag CD-20 en la superficie de las células B.
Epratuzumab es un Ac monoclonal IgG1 contra la molécula CD22. CD22 es una sialo-glicoproteína específica de las células B. Epratuzumab al unirse bloquea el receptor y acelera la apoptosis de las células B.
Ofatumumab: Ac monoclonal humano IgG1 anti CD-20, que induce la lisis de las células B. Actualmente está en ensayos clínicos en fase I/II (106).
Mavrilumabes: Ac monoclonal humanizado dirigido hacia el receptor a del GM-CSF (107).
Tabalumabes: un Ac monoclonal humanizado dirigido frente al BLyS (108).
Precauciones a seguir en los pacientes en tratamiento con terapia biológica (109).
Vacunación antineumocócica y antigripal (evitar vacunas con virus atenuados).
Vigilancia de la aparición de infecciones, citopenia grave y cáncer.
Exclusión de la existencia de tuberculosis activa o latente antes de iniciar una terapia biológica.
Precaución en pacientes con insuficiencia cardiaca congestiva moderada o grave.
Vigilancia de aparición de enfermedad desmielinizante.
Contraindicaciones para el uso de la terapia biológica:
Cáncer.
Enfermedad desmielinizante o neuritis óptica.
Citopenia grave.
Neumopatía intersticial nueva o agravamiento de previa u otros eventos graves relacionados con el fármaco.
Suspensión temporal si hay infección o en caso de cirugía mayor en período perioperatorio.
Valorar si hay embarazo o lactancia.
FARMACOGENÉTICA de la AR
Hay una gran variabilidad en la eficacia de los medicamentos así como en los efectos adversos de casi cualquier medicamento anti-reumático. A pesar de que los FAME detienen la progresión de la enfermedad en muchos pacientes (55%), hay un alto porcentaje de fracasos en la remisión de la enfermedad. Hoy por hoy, el establecer una terapia óptima para un paciente en particular es un proceso de ensayo y error, esto unido al alto coste de los fármacos biológicos, está conduciendo la investigación hacia la búsqueda de biomarcadores farmacogenéticos para optimizar el tratamiento de la AR (110).
REFERENCIAS
[1]. Klareskog, L.; Catrina, A. I.; Paget, S. Rheumatoid arthritis. Lancet 373, 659-672 (2009).
2. Scott, D. L.; Wolfe, F.; Huizinga, T. W. Rheumatoid arthritis. Lancet 376, 1094-1108 (2010).
3. Turesson, C. Extra-articular rheumatoid arthritis. Curr Opin Rheumatol 25, 360-366 (2013).
4. Alamanos, Y.; Voulgari, P. V.; Drosos, A. A. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Reumatology criteria: a systematic review. Semin Arthritis Rheum 36, 182-188 (2006).
5. Carmona, L.; Gonzalez-Alvaro, I.; Balsa, A.; Angel Belmonte, M.; Tena, X.; Sanmarti, R. Rheumatoid arthritis in Spain: occurrence of extra-articular manifestations and estimates of disease severity. Ann RheumDis62, 897-900 (2003).
6. Sociedad Española de Reumatología. Estudio EPISER: Prevalencia de las enfermedades reumáticas en la población española. Merck, Sharp &Dohme España; 2001.
7. Carbonell J.; Cobo T.; Balsa A.; Descalzo M. A.; Carmona L.The incidence of rheumatoid arthritis in Spain: results from a nationwide primary care registry. Rheumatology (Oxford) 47, 1088-1092 (2008).
8. Sanmartí, R.; Ruiz-Esquide, V.; Hernández, M. V. Rheumatoid Arthritis: A Clinical Overview of New Diagnostic and Treatment Approaches. Curr Top Med Chem 13, 698-704 (2013).
9. Van Vollenhoven, R. F. Rheumatoid arthritis in 2012: Progress in RA genetics, pathology and therapy. Nat Rev Rheumatol 9, 70-72(2013).
10. Carmona, L.; Cross, M.; Williams, B.; Lassere, M.; March, L. Rheumatoid arthritis. Best Pract Res Clin Rheumatol 24, 733-745 (2010).
11. MacGregor, A. J.; Snieder, H.; Rigby, A. S.; Koskenvuo, M.; Kaprio, J.; Aho, K. et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Reum 43, 30-37 (2000).
12. Bax, M.; Heemst, J. V.; Huizinga, T. W. J.; Toes, R. E. M. Genetics of rheumatoid arthritis: what have we learned?.Immunogenetics 63, 459-466 (2011).
13. Bowes, J.; Barton, A. Recent advances in the genetics of RA susceptibility. Rheumatology 47, 399-402 (2008).
14. Raychaudhuri,S.; Sandor, C.; Stahl, E. A.;Freudenberg, J.; Lee, H. S.; Jia, X. et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nature Genetics 44, 291-296 (2012).
15. Gibofsky, A. Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis.Am J Manag Care 18 (13 Suppl), S295-302 (2012).
16. Ding, B.; Padyukov, L.; Lundström, E.; Seielstad, M.; Plenge, R. M.; Oksenberg, J. R.; et al. Different patterns of associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in the extended major histocompatibility complex region. Arthritis Rheum 60, 30-38 (2009).
17. Varadé, J.; Figueredo, M. A.; Cano, S.; Fuentes, M.; Gómez de la Concha, E.; Fernández-Gutiérrez, B; et al. Association between shared epitope and anti-citrullinated peptide antibodies in rheumatoid arthritis: A systematic review and meta-analysis. Inmunología 30, 119-127 (2011).
18. O`Rielly, D. D.; Rahman, P. Pharmacogenetics of rheumatoid arthritis: Potential targets from susceptibility genes and present therapies. Pharmacogenomics and Personalized Medicine 3, 15-31 (2010).
19. Karlson, E. W.; Deane, K. Environmental and gene-environment interactions and risk of rheumatoid arthritis. Rheum Dis Clin North Am 38, 405-426 (2012).
20. Somers, E. C.; Thomas, S. L.; Smeeth, L.; Hall, A. J. Autoimmune diseases co-occurring within individuals and within families: a systematic review. Epidemiology 17,202-217(2006).
21. Goemaere, S.; Ackerman, C; Goethals, K.; De Keyser, F.; Van der Straeten, C.; Verbruggen; G.; et al. Onset of symptoms of rheumatoid arthritis in relation to age, sex and menopausal transition. J Rheumatol 17, 1620-1622 (1990).
22. Jacobsson, L. T.; Jacobsson, M.E.; Askling, J.; Knowler, W. C. Perinatal characteristics and risk of rheumatoid arthritis. BMJ 326, 1068-1069 (2003).
23. Feitsma, A. L.; Worthington, J.; van der Helm-van Mil, A. H.; Plant, D.; Thomson, W.; Ursum, J.; et al. Protective effect of noninherited maternal HLA-DR antigens on rheumatoid arthritis development. Proc Natl Acad Sci USA 104, 19966-19970 (2007).
24. Sugiyama, D.; Nishimura, K.; Tamaki, K.; Tsuji, G.; Nakazawa, T.; Morinobu, A.; et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Annals of the Rheumatic Diseases 69,70-81 (2010).
25. Klareskog, L.; Stolt, P.; Lundberg, K.; Kallberg, H.; Bengtsson, C.; Grunewald, J.; et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 54, 38-46 (2006).
26. Saevarsdottir, S.; Wedren, S.; Seddighzadeh, M.; Bengtsson, C.; Wesley, A.; Lindblad, S.; et al. Patients with early rheumatoid arthritis who smoke are less likely to respond to treatment with methotrexate and tumor necrosis factor inhibitors: observations from the Epidemiological Investigation of Rheumatoid Arthritis and the Swedish Rheumatology Register cohorts. Arthritis Rheum 63, 26-36 (2011).
27. Olsson, A.R.; Skogh, T.; Axelson, O.; Wingren, G. Occupations and exposures in the work environment as determinants for rheumatoid arthritis. Occup Environ Med 61, 233-238 (2004).
28. Jin, Z.; Xiang, C.; Cai, Q.; Wei, X.; He, J. Alcohol consumption as a preventive factor for developing rheumatoid arthritis: a dose-response meta-analysis of prospective studies. Ann RheumDis (2013). Disponible en: http://dx.doi.org/10.1136/annrheumdis-2013-203323
29. Agmon-Levin, N.;Theodor, E.;Segal, R. M.; Shoenfeld, Y.Vitamin D in systemic and organ-specific autoimmune diseases. Clin Rev Allergy Immunol 45, 256-266 (2013). Disponible en: http://dx.doi.org/10.1007/s12016-012-8342-y
30. Feser, M.; Derber, L. A.; Deane, K. D.; Lezotte, D. C.; Weisman, M. H.; Buckner, J. H.; et al. Plasma 25,OH vitamin D concentrations are not associated with rheumatoid arthritis (RA)-related autoantibodies in individuals at elevated risk for RA. J Rheumatol36, 943-946 (2009).
31. Calviello, G.; Su,H. M.; Weylandt, K. H.; Fasano, E.; Serini, S.; Cittadini, A. Experimental Evidence of 𝜔-3 Polyunsaturated Fatty Acid Modulation of Inflammatory Cytokines and Bioactive Lipid Mediators: Their Potential Role in Inflammatory, Neurodegenerative, and Neoplastic Diseases. Biomed Res Int 743171, (2013). Disponible en: http://dx.doi.org/10.1155/2013/743171
32. Miles, E. A.; Calder, P. C. Influence of marine n-3 polyunsaturated fatty acids on immune function and a systematic review of their effects on clinical outcomes in rheumatoid arthritis. Br J Nutr Jun 107 (Suppl S2), 171-184 (2012). Disponible en: http://dx.doi.org/10.1017/S0007114512001560.
33. Stamp, L. K.; James, M. J.; Cleland, L. G. Diet and rheumatoid arthritis: a review of the literature. Semin Arthritis Rheum 35,77-94 (2005).
34. Costenbader, K.H.; Kang, J.H.; Karlson, E. W. Antioxidant intake and risks of rheumatoid arthritis and systemic lupus erythematosus in women. Am J Epidemiol 172,205-216 (2010).
35. Scher, J.U.; Ubeda, C.; Equinda, M.; Khanin, R.; Buischi, Y.; Viale, A.; et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum 64, 3083-3094 (2012).
36. Mangat, P.; Wegner, N.; Venables, P.J.; Potempa, J. Bacterial and human peptidy largin in edeiminases: targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res Ther 12, 209(2010). Disponible en: http://arthritis-research.com/content/12/3/209.
37. Hitchon, C. A.; El-Gabalawy, H.S. Infection and rheumatoid arthritis: still an open question. CurrOpinRheumatol 23, 352-357 (2011).
38. Van den Oever, I. A.; van Sijl, A. M.; Nurmohamed, M. T.Management of cardiovascular risk in patients with rheumatoid arthritis: evidence and expert opinion. Ther Adv Musculoskelet Dis 5, 166-81 (2013). Disponible en: http://dx.doi.org/10.1177/1759720X13491025.
39. Ajeganova, S.; Andersson, M. L. &Hafström, I. Obesity is associated with worse disease severity in rheumatoid arthritis as well as with co-morbidities—a long-term follow-up from disease onset. Arthritis Care Res 65, 78-87 (2013). Disponible en:http://dx.doi.org/10.1002/acr.21710.
40. Wolfe, F.; Michaud, K. Effect of body mass index on mortality and clinical status in rheumatoid arthritis. Arthritis Care Res 64, 1471-1479 (2012).
41. Stavropoulos-Kalinoglou, A.; Metsios, G. S.; Panoulas, V. F.; Nightingale, P.; Koutedakis,Y.; Kitas, G. D. Anti-tumour necrosis factor α therapy improves insulin sensitivity in normal-weight but not in obese patients with rheumatoid arthritis. Arthritis Res Ther 14, R160 (2012).Disponible en: http://arthritis-research.com/content/14/4/R160.
42. Isaacs, J. D. The changing face of rheumatoid arthritis: sustained remission for all?. Nat Rev Inmunol10, 605-611 (2010).
43. Goronzy, J. J.; Weyand, C. M. Developments in the scientific understanding of rheumatoid arthritis. Arthritis Res Ther 11, 249 (2009). Disponible en: http://arthritis-research.com/content/11/5/249.
44. Colmegna, I.; Pryshchep, S.; Oishi, H.; Goronzy, J. J.; Weyand, C. M. Dampened ERK Signaling in Hematopoietic Progenitor Cells in Rheumatoid Arthritis. ClinImmunol143, 73-82 (2012). Disponible en: http://dx.doi.org/10.1016/j.clim.2012.01.007.
45. Van Boxel, J. A.; Paget, S. A. Predominantly T-cell infiltrate in rheumatoid synovial membranes. N Engl J Med 293, 517-520 (1975).
46. Edwards, J. C.; Cambridge, G.; Abrahams, V.M. Do self-perpetuating B lymphocytes drive human autoimmune disease? Immunology 97, 188-196(1999).
47. Astry, B.; Harberts, E.; Moudgil, K. D. A Cytokine-Centric View of the Pathogenesis and Treatment of Autoimmune Arthritis. J Interferon Cytokine Res 31, 927-940 (2011). Disponible en:http://dx.doi.org/10.1089/jir.2011.0094.
48. Sánchez-Ramón, S.; López-Longo, F. J.; Carreño, L. Interleucinas en la fisiopatología de la artritis reumatoide: más allá de las citocinas proinflamatorias. Reumatol Clin 6(S3), S20–S24 (2011).
49. Kazuo Okamoto, K.; Takayanagi, H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther13, 219 (2011). Disponible en http://arthritis-research.com/content/13/3/219.
50. Arboleya, L.; Castañeda, S. Osteoinmunología: el estudio de la relación entre el sistema inmune y el tejido óseo. ReumatolClin 9, 303-315 (2013).
51. Hamilton, J. A.; Tak, P. P. The dynamics of macrophage lineage populations ininflammatory and autoimmune diseases. Arthritis Rheum 60, 1210-1221(2009).
52. Chang, S.K; Gu, Z.; Brenner, M. B. Fibroblast-like synoviocytes in inflammatory arthritis pathology: the emerging role of cadherin-11. Immunol Rev 233,256-266 (2010).
53. Hueber, A. J.; Asquith, D. L.; Miller, A. M.; Reilly, J.; Kerr, S.; Leipe, J.; et al. Mast cells express IL-17A in rheumatoid arthritis synovium. J Immunol 184, 3336-3340 (2010).
54. Dieguez-Gonzalez, R.; Calaza, M.; Perez-Pampin, E.; Balsa, A.; Blanco, F. J.; Canete, J. D;et al. Analysis of TNFAIP3, a feedback inhibitor of nuclear factor-kappaB and the neighbor intergenic 6q23 region in rheumatoid arthritis susceptibility. Arthritis Res Ther11,R42 (2009).
55. Shegarfi, H.; Naddafi, F.; Mirshafiey, A. Natural Killer Cells and Their Role in Rheumatoid Arthritis: Friend or Foe?The Scientific World Journal 2012. Disponible en: http://dx.doi.org/10.1100/2012/491974
56. Yoshida, H.; Miyazaki, Y. Regulation of immune responses by interleukin-27. Immunol Rev 226, 234-247(2008).
57. Stack, R.J.; Sahni, M.; Mallen, C. D.; Raza K. Symptom complexes at the earliest phases of rheumatoid arthritis: A synthesis of the qualitative literature. Arthritis Care Res 2013. Disponible en: http://dx.doi.org/10.1002/acr.22097
58. Ek, M.; Engblom, D.; Saha, S.; Blomqvist, A.; Jakobsson, P. J.; Ericsson-Dahlstrand, A. Inflammatory response: pathway across the blood-brain barrier. Nature 410, 430-431(2001).
59. Moreland, L. W.; Genovese, M. C.; Sato, R.; Singh, A. Effect of etanercept on fatigue in patients with recent or established rheumatoid arthritis. Arthritis Rheum 55, 287-293 (2006).
60. Kuriya, B.; Villeneuve, E.; Bombardier, C. Diagnostic and prognostic value of history-taking and physical examination in undifferentiated peripheral inflammatory arthritis: a systematic review. J Rheumatol Suppl, 87, 10-14 (2011).
61. Arnett, F. C.; Edworthy, S. M.; Bloch, D. A.; McShane, D. J.; Fries, J. F.; Cooper, N. S.; et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31, 315-324 (1988).
62. Harrison, B. J.; Symmons, D. P.; Barrett, E.M.; Silman, A. J. The performance of the 1987 ARA classification criteria for rheumatoid arthritis in a population based cohort of patients with early inflammatory polyarthritis. American Rheumatism Association.J Rheumatol25, 2324-2330 (1998).
63. Finckh, A.; Liang, M.H.; van Herckenrode, C.M.; de Pablo, P. Long-term impact of early treatment on radiographic progression in rheumatoid arthritis: A meta-analysis. Arthritis Rheum55, 864-872 (2006).
64. Schneider, M.; Krüger K. Rheumatoid arthritis—early diagnosis and disease management. Dtsch Arztebl Int 110, 477-484 (2013).
65. Kay, J.; Upchurch, K. S. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology (Oxford) 51 (Suppl6), vi5-vi9 (2012). Disponible en http://dx.doi.org/10.1093/rheumatology/kes279.
66. Van der Linden, M.P.; Batstra, M.R.; Bakker-Jonges, L.E.; Detert, J.; Bastian, H.; Scherer, H.U.; et al. Toward a data-driven evaluation of the 2010 American College of Rheumatology / European League Against Rheumatism criteria for rheumatoid arthritis: is it sensible to look at levels of rheumatoid factor? Arthritis Rheum 63, 1190-1199 (2011).
67. Humphreys, J.H.; Symmons, D.P. Postpublication validation of the 2010 American College of Rheumatology/European League Against Rheumatism classification criteria for rheumatoid arthritis: where do we stand? Curr Opin Rheumatol 25, 157-163 (2013).
68. Hulsemann, J. L.; Zeidler, H. Undifferentiated arthritis in an early synovitis out-patient clinic. Clin Exp Rheumatol 13, 37-43, (1995).
69. Mochan E, Ebell M. H. Predicting rheumatoid arthritis risk in adults with undifferentiated arthritis. Am Fam Physician 77, 1451-1453 (2008).
70. Lamana, A.; Balsa, A.; Rueda, B.; Ortiz, A. M.; Nuño, L.; Miranda-Carus, M. A.; et al. The TT Genotype of the STAT4 rs7574865 Polymorphism Is Associated with High Disease Activity and Disability in Patients with Early Arthritis. PLoS ONE 7, e43661 (2012). Disponible en http://dx.doi.org/10.1371/journal.pone.0043661
71. Balsa A, Cabezón A, Orozco G, Cobo, T.; Miranda-Carus, E.; López-Nevot, M. A.; et al. Influence of HLA DRB1 alleles in the susceptibility of rheumatoid arthritis and the regulation of antibodies against citrullinated proteins and rheumatoid factor. Arthritis Res Ther 12, R62 (2010). Disponible en: http://arthritis-research.com/content/12/2/R62
72. Lundberg, K.; Bengtsson, C.;Kharlamova, N.;Reed, E.;Jiang, X.;Kallberg, H.; et al. Genetic and environmental determinants for disease risk in subsets of rheumatoid arthritis defined by the anticitrullinated protein/peptide antibody fine specificity profile. Ann Rheum Dis 72, 652-658 (2013). Disponible en: http://dx.doi.org/10.1136/annrheumdis-2012-201484
73. Ravelli, A.; Felici, E.; Magni-Manzoni, S.; Pistorio, A.; Novarini, C.; Bozzola, E.; et al. Patients with antinuclear antibody-positive juvenile idiopathic arthritis constitute a homogeneous subgroup irrespective of the course of joint disease. Arthritis Rheum 52, 826-832 (2005).
74. Yuasa, S.; Yamaguchi, H.; Nakanishi, Y.; Kawaminami, S.; Tabata, R.; Shimizu, N.; et al. Treatment responses and their predictors in patients with rheumatoid arthritis treated with biological agents. J Med Invest 60, 77-90 (2013).
75. Willemze, A.;Toes, R. E.;Huizinga, T. W.;Trouw, L. A.New biomarkers in rheumatoid arthritis. Neth J Med 70, 392-399 (2012).
76. Disponible en: http://www.omeract.org/
77. Welsing, P. M.; van Gestel, A. M.; Swinkels, H. L.; Kiemeney, L. A.; van Riel, P.L. The relationship between disease activity, joint destruction, and functional capacity over the course of rheumatoid arthritis. Arthritis Rheum 44, 2009-2017 (2001).
78. Anderson, J.K.; Zimmerman, L.; Caplan, L. ; Michaud, K. Measures of rheumatoid arthritis disease activity: Patient (PtGA) and Provider (PrGA) Global Assessment of Disease Activity, Disease Activity Score (DAS) and Disease Activity Score with 28-Joint Counts (DAS28), Simplified Disease Activity Index (SDAI), Clinical Disease Activity Index (CDAI), Patient Activity Score (PAS) and Patient Activity Score-II (PASII), Routine Assessment of Patient Index Data (RAPID), Rheumatoid Arthritis Disease Activity Index (RADAI) and Rheumatoid Arthritis Disease Activity Index-5 (RADAI-5), Chronic Arthritis Systemic Index (CASI), Patient-Based Disease Activity Score With ESR (PDAS1) and Patient-Based Disease Activity Score without ESR (PDAS2), and Mean Overall Index for Rheumatoid Arthritis (MOI-RA).Arthritis Care Res 63(Suppl 11), S14-36 (2011).
79. Prevoo, M.L.; van 't Hof, M.A.; Kuper, H.H.; van Leeuwen, M.A.; van de Putte, L.B.;van Riel, P.L. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis.Arthritis Rheum 38, 44-48 (1995).
80. Hochberg, M. C.; Chang, R. W.; Dwosh, I.; Lindsey, S.; Pincus, T.; Wolfe F. The AmericanCollege of Rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum 35, 498-502 (1992).
81. Emery, P.; Breedveld, F.C.; Hall, S.; Durez, P.; Chang, D.J.; Robertson, D.; et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet 372, 375-382 (2008).
82. Ma, M.H.; Scott, I.C.; Kingsley, G.H.; Scott, D.L. Remission in early rheumatoid arthritis. J Rheumatol 37, 1444-1453 (2010).
83. Jayakumar, K.; Norton, S.; Dixey, J.; James, D.; Gough, A.; Williams, P.; et al. Sustained clinical remission in rheumatoid arthritis: prevalence and prognostic factors in an inception cohort of patients treated with conventional DMARDS. Rheumatology (Oxford) 51, 169-175 (2012).
84.Shammas, R.M.; Ranganath, V. K.; Paulus, H.E. Remission in rheumatoid arthritis. CurrRheumatol Rep 12, 355-362 (2010).
85. Brown, A.K.; Conaghan, P.G.; Karim, Z.; Quinn, M.A.; Ikeda, K.; Peterfy, C.G.; et al. An explanation for the apparent dissociation between clinical remission and continued structural deterioration in rheumatoid arthritis.ArthritisRheum 58, 2958-2967 (2008).
86. Maesea, J.; García De Yébenes, M. J.; Carmona, L.; Hernández-García, C.; el Grupo de Estudio emAR II. Estudio sobre el manejo de la artritis reumatoide en España (emAR II). Características clínicas de los pacientes. Reumatol Clin 8, 236-242 (2012).
87. Niedermann, K.; de Bie, R. A.; Kubli, R.; Ciurea, A.; Steurer-Stey, C.; Villiger, P. M.; et al. Effectiveness of individual resource-oriented joint protection education in people with rheumatoid arthritis: a randomized controlled trial. Patient Educ Couns 82, 42-48 (2011).
88. Steultjens, E. M.; Dekker, J.; Bouter, L. M.; van Schaardenburg, D.; van Kuyk, M. A.; Van den Ende, C. H. Occupational therapy for rheumatoid arthritis. Cochrane Database Syst Rev1,(2004). Disponible en: http://dx.doi.org/10.1002/14651858.CD003114.pub2.
89. Hewlett, S.; Ambler, N.; Almeida, C.; Cliss, A.; Hammond, A.; Kitchen, K.; et al. Self-management of fatigue in rheumatoid arthritis: a randomised controlled trial of group cognitive-behavioural therapy. Ann Rheum Dis70, 1060-1067 (2011).
90. Hagen, K. B.; Dagfinrud, H.; Moe, R. H.; Østerås, N.; Kjeken, I.; Grotle, M.; et al. Exercise therapy for bone and muscle health: an overview of systematic reviews. BMC Medicine 10, (2012). Disponible en: http://www.biomedcentral.com/1741-7015/10/167.
91. Tokuyama, S.; Nakamoto, K. Current Topics Lipid Mediators and Pain Signaling Unsaturated Fatty Acids and Pain. BiolPharm Bull 34, 1174-1178 (2011).
92. Yeoh, N.; Burton, J. P.;Suppiah, P.;Reid, G.;Stebbings, S.The role of the microbiome in rheumatic diseases. Curr Rheumatol Rep 15, 314 (2013). Disponible en: http//dx.doi.org/ 10.1007/s11926-012-0314-y.
93. Saag, K. G. Short-term and Long-term Safety of Glucocorticoids in Rheumatoid Arthritis. Bull NYU HospJt Dis 70(Suppl 1),S21-25 (2012).
94. Cheng, O. T.; Souzdalnitski, D.;Vrooman, B.; Cheng, J. Evidence based knee injections for the management of arthritis. Pain Med June 13, 740-753 (2012).
95. Baggott, J. E.;Morgan, S. L. Methotrexate catabolism to 7-hydroxy methotrexate in rheumatoid arthritis alters drug efficacy and retention and is reduced by folic acid supplements. Arthritis Rheum 60, 2257-2261 (2009).
96. O'Dell, J. R.; Mikuls, T. R.; Taylor, T. H.; Ahluwalia, V.; Brophy, M.; Warren, S. R.; et al. Therapies for active rheumatoid arthritis after metotrexate failure. N Engl J Med 369, 307-318 (2013).
97. Smolen, J. S.; Aletaha, D. Forget personalized medicine and focus on abating disease activity. Ann Rheum Dis 72, 3-6 (2013).
98. Rosman, Z.; Shoenfeld, Y.; Zandman-Goddard, G. Biologic therapy for autoimmune diseases: an update. BMC Medicine11, 88 (2013). Disponible en: http://www.biomedcentral.com/1741-7015/11/88
99. Malviya, G.; Salemi, S.; Laganà, B.; PicchiantiDiamanti, A.; D’Amelio, R.; Signore, A. Biological Therapies for Rheumatoid Arthritis: Progress to Date. Bio Drugs 27, 329-345 (2013).
100. Smolen, J. S.; Emery, P. Infliximab: 12 years of experience. Arthritis Res Ther13 (Suppl 1), S2 (2011). Disponible en: http://arthritis-research.com/supplements/13/S1/S2.
101. Al-Shakarchi, I.; Gullick, N.J.; Scott, D. L. Current perspectives on tocilizumab for the treatment of rheumatoid arthritis: a review. Patient Preference and Adherence 7, 653-666 (2013).
102. Smolen, J. S.; Schoels, M. M.; Nishimoto, N.; Breedveld, F.C.; Burmester, G.R.; Dougados, M. et al. Consensus statement on blocking the effects of interleukin-6 and in particular by interleukin-6 receptor inhibition in rheumatoid arthritis and other inflammatory conditions. Ann Rheum Dis 72, 482-92 (2013). Disponible en: http://dx.doi.org/10.1136/annrheumdis-2012-202469.
103. Athanasakis, K.; Petrakis, I.; Kyriopoulos, J. Investigating the Value of Abatacept in the Treatment of Rheumatoid Arthritis: A Systematic Review of Cost-Effectiveness Studies. ISRN Rheumatology, 2013. Disponible en: http://dx.doi.org/10.1155/2013/256871.
104. Kawalec, P.; Mikrut, A.; Wiśniewska, N.; Pilc. A. The effectiveness of tofacitinib, a novel Janus kinase inhibitor, in the treatment of rheumatoid arthritis: a systematic review and meta-analysis. Clin Rheumatol 32, 1415-1424 (2013).
105. Jin, X.; Ding, C. Belimumab - An anti-BLyS human monoclonal antibody for rheumatoid arthritis. Expert Opinion on Biological Therapy 13, 315-322 (2013).
106. Kurrasch, R.; Brown, J.C.; Chu, M.; Craigen, J.; Overend, P.; Patel, B.; et al. Subcutaneously administered ofatumumab in rheumatoid arthritis: a phase I/II study of safety, tolerability, pharmacokinetics, and pharmacodynamics. J Rheumatol 40, 1089-1096 (2013).
107. Combe, B.; van Vollenhoven, R. Novel targeted therapies: the future of rheumatoid arthritis? Mavrilumab and tabalumab as examples. Ann Rheum Dis 72, 1433-1435 (2013).
108. Genovese, M.C.; Fleischmann, R.M.; Greenwald, M.; Satterwhite, J.; Veenhuizen, M.; Xie, L.; et al. Tabalumab, an anti-BAFF monoclonal antibody, in patients with active rheumatoid arthritis with an inadequate response to TNF inhibitors. Ann RheumDis72, 1461-1468 (2013).
109. Gómez Reino, J.; Loza, E.; Andreu, J. L.; Balsa, A.; Batlle, E.; Cañete, J. D.; et al. Consenso SER sobre la gestión de riesgo del tratamiento con terapias biológicas en pacientes con enfermedades reumáticas. Reumatol Clin 7, 284-298 (2011).
110. Umićević Mirkov, M.; Coenen, M.J. Pharmacogenetics of disease-modifying antirheumatic drugs in rheumatoid arthritis: towards personalized medicine. Pharmacogenomics 14,425-444 (2013).