REVISIÓN |
Class I phosphoinositide 3-kinases in immunity: Effect of inhibitors in immune and autoimmune reactions
José María Rojo1,2, Pilar Portolés3*
1Departamento de Medicina Celular y Molecular, Centro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu, 9, 28040, Madrid, Spain. 2Acádemico Correspondiente de la Real Academia Nacional de Farmacia. 3Unidad de Inmunología Celular, Centro Nacional de Microbiología, Instituto de Salud Carlos III; Ctra. Majadahonda-Pozuelo km. 2, Majadahonda, 28220-Madrid, Spain.
*e-mail: pportols@isciii.es
Recibido el 9 de enero de 2014 - An. Real Acad. Farm. Vol. 80, Nº 1 (2014), pág. 91-125
abstract
Class I phosphoinositide-3 kinases (PI3Ks) generate PtdIns(3,4,5)P3 to activate cell signaling cascades essential to cell growth, differentiation and survival, and are essential to the function of innate and adaptive immunity. The generation of a vast array of newly developed PI3K inhibitors to treat cancer poses the question of their use in the modulation of pathological immune reactions like autoimmune diseases, or the effect of these drugs in the anti-tumor immune reactions. Here, the role of PI3K in adaptive immune reactions and data concerning the use of inhibitors to control immune responses are reviewed. |
Keywords: PI3K; PI3K inhibitors; Immune responses.
resumen
Las fosfatidilinositol 3-cinasas de clase I en la inmunidad: Efecto de inhibidores en respuestas inmunes y autoinmunes
Las fosfatidilinositol 3-cinasas (PI3K) de clase I dan lugar a fosfolípidos trifosforilados (PtdIns(3,4,5)P3) que son clave en las señales de crecimiento, diferenciación y la supervivencia de las células y son esenciales para el funcionamiento de la inmunidad innata y adaptativa. Los nuevos inhibidores de PI3K generados para el tratamiento de tumores pueden ser útiles en inmunoterapia, especialmente en enfermedades autoinmunes, y ha de investigarse su impacto en la inmunidad anti tumoral. Se revisa el papel de las PI3K de clase I en las respuestas inmunes adaptativas, y los datos conocidos relativos al efecto de inhibidores en respuestas inmunes adaptativas. |
Palabras clave: PI3K; Inhibidores de PI3K; Respuesta inmunitaria.
1. INTRODUCtioN
This review focuses on the use and possible applications of phosphoinositide-3 kinase (PI3K)-specific inhibitors to modulate immune responses, with an emphasis on T lymphocyte-dependent adaptive responses.
Phosphoinositide-3 kinases (PI3Ks) are enzymes that phosphorylate the OH- group at the D3-position of the inositol ring of inositol-containing lipids (PtdIns) located in inner leaflet of membrane bilayers. In this way, they generate intracellular phosphorylated inositol lipids (PtdInsP) that serve to anchor cytosolic enzymes facilitating interaction with their substrates and/or their activation, initiating different cell signaling cascades (Figure 1) (reviewed in (1-5) ). This is mediated by membrane translocation of different effector proteins that possess domains, like the Pleckstrin homology domain (PH), the Phox homology domain (PX) or the FYVE domain, specific for distinct phosphorylated inositides (Figure 1). These effector proteins are involved in the regulation of many essential cell functions including cell survival, growth, and proliferation. Not surprisingly, different signals essential to the function of cells of the immune system activate PI3K activity. For instance, in lymphocytes PI3K activity is enhanced upon antigen activation, engagement of costimulatory molecules, binding of cytokines or quimiokines to their receptors, or integrin-mediated adhesion. Thus, PI3Ks are prime targets for immunomodulatory strategies.
Differences in protein structure, regulation of activity, and lipid substrate preference define three different classes of PI3K, namely class I, class II and class III PI3K (Figure 1, 2). In mammals there are eight different PI3K isoforms. Of them, four are class I PI3Ks (p110α, p110β, p110γ and p110δ), three belong to class II PI3K (PI3K-C2α, PI3K-C2β, PI3K-C2γ) and the vacuolar sorting protein 34 (VPS34) is the only class III PI3K. Class I are PI3Kinases specific for PtdIns(4,5)P2 and play a major role in signal transduction induced by receptors that activate protein tyrosine kinases (class IA PI3K) or by receptors coupled to small GTPases (class IB PI3K). Class II and III PI3Kinases phosphorylate PtdIns. Class II have a role in signal transduction, but many aspects of their biology are not well known; Class III have a role in vesicle trafficking.

Figure 1.- A summary of phosphoinositide phosphorylation/dephosphorylation steps in the cell membrane is shown. These phosphoinositides recruit proteins endowed with phosphorylated inositol lipid-binding protein domains that serve to cell signaling. Phosphorylation and dephosphorylation steps are indicated by red and blue arrows, respectively; steps where PI3K kinases intervene are shadowed in red; Inositol phosphate phosphatases are shadowed in blue; effector protein domains binding to different phosphorylated inositol lipids are in yellow. Abbreviations: FAB1: PtdIns(3)P 5-kinase; FYVE: Fab 1 YOTB Vac 1 and EEA1 domain; INPP4: Inositol polyphosphate 4-phosphatase; INPP5: Inositol polyphosphate 5-phosphatase; MTM: Myotubularin; PH: Plecstrin homology domain; PI3K: Phosphoinositide-3 kinase; PLC: Phospholipase C; PROPPIN: b-propeller that bind phosphoinositide species domain; PtdInsP: Phosphorylated inositol lipids; PTEN: Phosphatase and Tensin Homologue; PX: Phox homology domain; SHIP: SH2 domain-containing inositol 5’-phosphatase.
2. Class I, II, III PI3K, and PI3K-like kinases
Of interest to the development and effect of PI3K-specific inhibitors, PI3Ks have sequence similarity with a number of other related serine and threonine protein kinases collectively known as “PI3K-like protein kinases”, or PIKK. These PIKK play a role in the cellular response to stresses like DNA damage or replication block, mRNA splicing errors and nutrient deprivation. Importantly, PIKK include one of the main downstream effectors of class I PI3K activation in lymphocytes, namely the mechanistic target of the immunosuppressant Rapamycin (mTOR), but also the ataxia-telangiectasia mutated protein (ATM), the ataxia- and Rad3-related protein (ATR), and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) (6,7) . DNA-PK is required for the activation of the non-homologous end-joining pathway to repair double-strand DNA breaks induced by ionizing radiation. Furthermore, DNA-PK activates protein kinase B (Akt), another primer target of class I PI3K, and is particularly important to lymphocyte biology as its mutation is a cause for severe combined immunodeficiency (8) . Structural homology among PI3K isoforms, or between PI3K and PIKK (6) has the consequence that frequently, PI3Kinase inhibitors are specific for more than one isoform, or that PI3K inhibitors primarily aimed at inhibiting class I PI3Kinases significantly inhibit PIKK like mTOR or DNA-PK (9) .
3. Class IA and Class IB PI3Kinases: Structure and activation, control by phosphatases
Class I PI3Kinases are heterodimers formed by one catalytic of 110 kDa and one regulatory subunit of variable size (Figure 2). According to the nature of the regulatory subunit they bind to, and the mode they are classically activated, class I PI3Kinases are further divided into class IA (Figure 2A and B) and class IB (Figure 2C). The former (catalytic subunits p110a, p110b, and p110d) bind to any of five regulatory subunit isoforms (p85a, p55a, p50a, p85b, and p55g) all possessing two SH2 domains separated by an alpha-helix inter-SH2 domain (iSH2) that interacts with catalytic subunits; Class IA catalytic subunits are characteristically activated through tyrosine kinase-dependent mechanisms. Phosphorylation of the Tyr residues of Tyr-x-x-Met is the initial step inducing binding of SH2 domains that are conserved in all class IA regulatory isoforms. The p85, but not the p50 or p55 regulatory isoforms have additional SH3, proline rich and BCR-homology GTPase activation domain (BH-domain) that might mediate binding to small G proteins of the Rho/Rac/cdc42 family.
On the other hand, class IB catalytic subunit p110g binds to regulatory subunits p101 and p87 (Figure 2C). Subunit p101 is the main subunit in most tissues; p101 and p85 differentially help in G protein–coupled receptor (GPCR)-induced PI3K activation, such that interaction of p101 with the Gβγ chains is strong and efficient in recruitment of p110γ to membranes whereas p87 interacts weakly with Gβγ chains and recruitment of p110γ needs additional interactions with Ras-GTP (10) .
Class I catalytic subunits are structured in domains with distinct function (Figure 2A-C); these include a N-terminal adaptor binding domain (ABD) that constitutively bind regulatory subunits, a Ras-binding domain, the C2 and helical domains that regulate the association with regulatory subunits, and one C-terminal kinase domain. Interactions of class IA p110a and the iSH2 and amino-terminal domain of regulatory p85a subunits have been determined in detail (11,12) . In the steady state, the p110-p85 heterodimers have low enzymatic activity; however, binding of SH2 domains to phosphorylated peptides disrupt the inhibitory interactions including the one between the p85 nSH2 domain and the helical domain of p110a (12,13) . Given the inhibitory nature of the regulatory subunits, it should be noted that there are clear differences among catalytic isoforms concerning their interaction strength with regulatory subunits (14) . In addition, at least in certain cases Ras-family proteins might play an active role in recruiting to membranes catalytic class IA (15) and class IB subunits (10) .
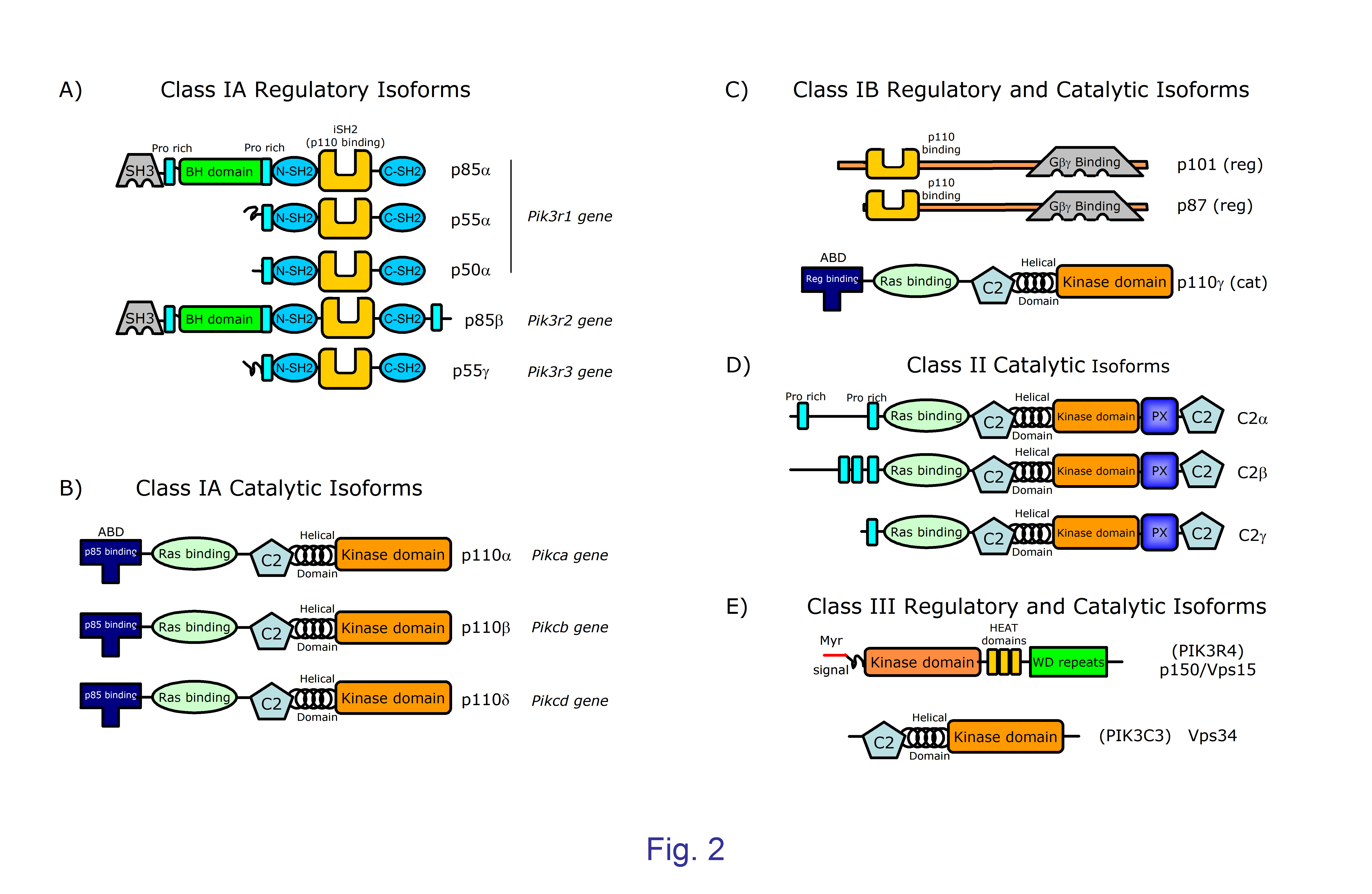
Figure 2. Structure of the catalytic and regulatory subunits of mammalian PI3K classes and subclasses. Size in kDa is indicated in the name of class I PI3K, all proteins are represented at the same approximate scale. PI3K catalytic subunits contain a common core of one C2 domain, one helical domain, and one catalytic kinase domain. Class IA and IB catalytic subunits (a, b, d, and g) are the product of single genes ; they have two domains N-terminal to the core, namely an adaptor binding domain (ABD) that binds to regulatory subunit, and one Ras-binding domain where binding of Ras family proteins activate the kinase activity. A; B) Class IA catalytic subunits (a, b, and d) associate with any class IA regulatory subunits encoded by three different genes. Pik3r1 can produce three different proteins (p85a, p55a and p50a) sharing one Pro-rich region, plus one N-terminal and one C-terminal SH2 domains separated by and inter-SH2 domain (iSH2) that binds the ABD domain in the catalytic a, b, and d subunits. The p85a subunit has one N-terminal SH3, one Proline-rich, and one BH (BCR homology) domain; the p85b subunit coded by the Pik3r2 gene is similar to p85a subunit but has an additional c-terminal Pro-rich region. The p55g subunit encoded by Pik3r3 and p55a have similar structures. C) Class IB catalytic subunits (p110g) bind to p87 or p101 class IB regulatory subunits endowed with domains able of associating to the Ga and Gb subunits of heterotrimeric Guanine nucleotide-binding proteins (G proteins) that initiate signals delivered by G-protein coupled receptors (GPCR). D) Class II PI3Ks do not have regulatory subunits, and seem to be constitutively bound to intracellular membranes. They have a role in different cell functions including cell migration, exocytosis, and apoptosis, but the precise mechanisms involved are not clear. The Class III catalytic subunit Vps34 (Vacuolar protein sorting 34, also termed PIK3C3) is part of a heterodimer with the myristoylated protein Vps15, that is located in the cell membranes and form larger multi-protein complexes depending on the particular vesicle traffic process considered (autophagy, phagocytosis, endosome traffic). Vps15 has a kinase domain probably inactive, HEAT domains containing anti-parallel a-helices involved in protein-protein interactions; and WD repeats that serve as scaffolds for interaction with other proteins.
Regulatory subunits inhibit the catalytic activity of the p110 subunits but also prevent their degradation. Activation of PI3K begins upon recruitment of the enzyme complex through regulatory subunits to the inner side of membrane bilayers where their substrate is located, and where interaction with the negatively charged surface through positively charged aminoacid residues further stabilizes its location. This is followed by interaction with and activation of the catalytic subunits by small GTPases of the Ras family, with a clear selectivity of the different catalytic subunits for activation by distinct Ras family members (16) .
The signaling outcome of PI3K is also controlled by the activity of PtdIns(3,4,5)P3 phosphatases that are essential to correct lymphocyte function (17-19) (Figure 1, 3). The PtdIns(3,4,5)P3 3-phosphatase Phosphatase and Tensin Homologue (PTEN) controls PtdIns(3,4,5)P3 levels by generating PtdIns(4,5)P2; PTEN is particularly important to control basal levels of PtdIns(3,4,5)P3. The PtdIns(3,4,5)P3 5-phosphatases SHP-1 and SHP-2 (SH2 domain-containing Inositol Phosphatase-1 and -2) dephosphorylate PtdIns(3,4,5)P3 levels yielding PtdIns(3,4)P2. Interestingly, SHP-1, 2 bind to surface molecules of lymphocytes that are essential to negatively control immune responses including CTLA-4, PD-1, and BTLA (17) .
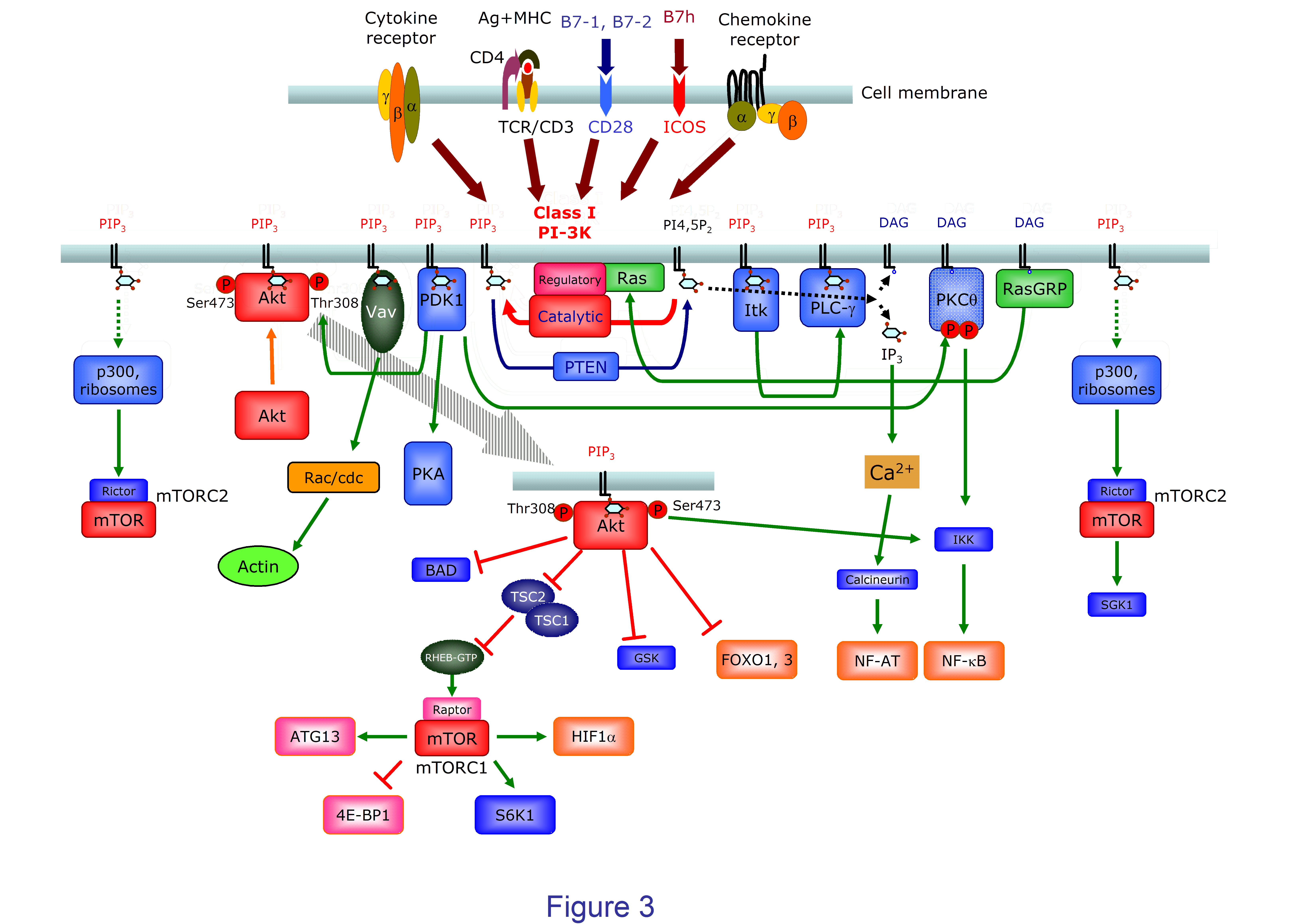
Figure 3. A diagram of class I PI3K-mediated signaling in T (CD4+) lymphocytes. Class IA PI3K are recruited to the cell membrane and activated upon binding of ligands for cytokine and antigen receptors or CD28 family costimulator molecules like CD28 or ICOS. Class IB subunits are characteristically recruited and activated upon binding of chemokines to their G protein-coupled receptors. Enhanced levels of PtdIns(3,4,5)P3 (PIP3) favor the recruitment and activation of proteins containing PH domains including Ser and Thr kinases like PDK1 and Akt, Tec family protein Tyr kinases (Itk), phospholipases like PLC-g, or GEF like Vav. Akt is activated by phosphorylation of Thr308 and Ser473 by PIP3–dependent activated PDK1 and mTORC2, respectively. Akt has an array of different targets involved in many cell functions. One major effector of Akt is the mTORC1 complex containing the Ser/Thr kinase mTOR and Raptor. This is achieved by inactivation/phosphorylation of the tuberous sclerosis 1 and 2 complex (TSC1-TSC2) that blocks RHEB (Ras homologue enriched in brain), a small GTPase and mTOR activator. Through phosphorylation of other substrates like ATG13 (autophagy-related protein 13), S6K, 4E-BP1, or HIF-1a, mTORC1 controls important cell processes including autophagy or cell metabolism. Akt also regulate apoptosis through Bcl-2-associated death promoter (BAD) protein phosphorylation, the transcription of genes controled by FOXO transcription factors, or Glycogen synthase kinase (GSK) 3-mediated effects on cell cycle and metabolism. mTORC2 and PDK1 also phosphorylate the PKCq activated by diacylglicerol (DAG), or Serum- and glucocorticoid-inducible kinase (SGK) 1. PLC-g is recruited to PI3P3 containing membranes to generate IP3 and DAG; IP3 induces enhanced cytoplasmic Ca2+ levels and activation of NF-AT transcription factors. DAG activates not only NF-kB through PKCq, but RasGRP that activates the Ras proteins, thus enhancing PI3K and other Ras-dependent signals including the MAPK pathway.
One, then, faces a situation where class I PI3K, an enzyme essential to many cell functions is regulated at many levels, particularly: i) by the expression levels of each regulatory and catalytic isoform of PI3K, of the different PtdIns(3,4,5)P3 phosphatases, and the different activator and effector proteins of PI3K; and ii) by the nature of the signals that activate PI3K activity in different cells and conditions. The final outcome of the cell response will depend on the integration of all these factors. Furthermore, differences among different cells and tissues concerning these factors allow for distinct pharmacological effects of PI3Kinase inhibitors and a selective intervention in different pathological situations.
As a general rule, the expression of class IA p110a and p110b is broad, whereas class IA p110d and class IB p110g are mainly expressed by cells of hematopoietic origin. Among class I PI3K, lymphocytes express readily detectable amounts of class IA p110d and class IB p110g catalytic subunits that are characteristic of leukocytes. Interestingly, in T lymphocytes class IA p110a is expressed at levels similar to p110d, but expression of p110b is low (14) . The particular function for the different class IA regulatory subunit isoforms is not clearly established. Among class IA regulatory subunits, T lymphocytes express low amounts of p85b and p55g chains ( (14) , and unpublished data), so p85a and its splicing variants p55a and p50a are the main regulatory subunits (14) . There are clear variations in the abundance of these isoforms in resting and activated T cells. Thus, resting CD4+ T lymphocytes have similar levels of p85a and p50a, but the levels of p50a is strongly reduced upon activation; low levels of p50a are also common in T cell lines (14) . In turn, p50a subunits bind better than p85a to phosphorylated Tyr-x-x-Met motifs (14) and are preferentially recruited to immunological synapses together with ICOS (20) . However, the functional meaning of these differences is far from clear.
4. Class IA and Class IB PI3Kinases: Main molecular mediators
In lymphocytes class I PI3Kinases are activated through different receptors ( (21,22) , Figure 3). The activity of Src and Syk tyrosine kinases is triggered by ligand binding to antigen receptor complexes (BCR in B lymphocytes, TCR/CD3 in T lymphocytes). This activation leads to phosphorylation of adaptor proteins like LAT and TRIM in T cells or BCAP in B cells. Class IA PI3Kinases are typically recruited to membranes and activated upon phosphorylation of Y-x-x-M motifs in these proteins (i.e. in TRIM) or by means of adaptor proteins like Grb-2 or Cbl. Co-stimulatory molecules like CD28 and ICOS in T cells and CD19 in B cells also possess Y-x-x-M motifs that can be phosphorylated to directly recruit PI3Kinases. Cytokine receptors indirectly induce recruitment and activation of class IA PI3K by means of different adaptor proteins. For instance, in IL-2 signaling IL-2 receptors (IL-2R) recruits class IA PI3K upon phosphorylation of Tyr338 of the b chain that allows binding of the protein adaptors SHP2 and Gab2, as well as the Shc and Grb adaptors that facilitate the formation of Grb-Gab2 o Grb-2-SHPS. On the other hand, the PI3Kinase class IB p110g catalytic subunits are activated by the bg subunits of G-coupled protein chemokine receptors having seven trans-membrane peptide sequences. However, this separation among PI3K subclasses is not absolute, as on one hand class IA p110b signaling through G-coupled receptors has been observed ( (23) , reviewed in (5) ), and in T lymphocytes tyrosine kinases like Lck and ZAP-70 can associate p110g class IB subunits upon antigen receptor activation (24) . This is further complicated in lymphocytes, where p110g mediate PI3K-dependent chemokine signals in T lymphocytes, but p110d perform the same functions in B lymphocytes by mechanisms not well understood (25,26) , but that might involve tyrosine kinases or Ras members activated by quimiokines (5) .
Upon PI3K activation PtdIns(3,4,5)P3 is generated that can be dephosphorylated by the action of SHIP phosphatases to yield PtdIns(3,4)P2. Both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 recruit proteins with plectstrin homology (PH) domains to cell membranes, usually the plasma membrane. Recruited proteins can then be activated to initiate signaling cascades. These proteins can vary, as PH domains have certain selectivity concerning their interaction strength with these phosphoinositides. In lymphocytes they include serine threonine kinases like the phosphoinositide-dependent kinase 1 (PDK1) or Akt (PKB), Tec family tyrosine kinases like Itk (in T lymphocytes) or Btk (in B lymphocytes), adaptor proteins like GAB2, and small GTPase positive (Guanine nucleotide exchange factors (GEF) like Vav) or negative (GTPase activating factors (GAP)) regulatory proteins.
4.a. Targets of PI3K: PDK1, Akt and mTOR.
In many cell types and in lymphocytes too, the serine threonine kinase Akt plays a key mediator role in PI3K activated signals (3,18,27-29) . Membrane-recruited Akt is activated upon phosphorylation in residue Thr308 by PDK1 that is also recruited through PH domains and activated by tyrosine kinases of the src-family kinases like Lck (30) . Akt P-Thr308 then phosphorylates substrates like Caspase 9, BAD or IKKa to prevent apoptosis, GSK3b to favour proliferation, and the mTORC1 complex (the Rapamycin sensitive complex of mechanistic target of Rapamycin (mTOR) containing Raptor) or its negative regulator TSC1/2 to favour protein synthesis and cell growth. Akt is fully activated by phosphorylation of Ser423 by kinases generically termed PDK2. The main PDK2 is mTORC2, the Rapamycin-insensitive complex of mTOR containing Rictor. mTORC2 activation is also PI3K dependent and involves its association with ribosomes, yet the exact mechanisms are not known (29) . At least another well established PDK2 activity for Ser423 Akt phosphorylation is mediated by the DNA-PK (7,31,32) . Doubly phosphorylated Akt P-Thr308Ser423 efficiently phosphorylates forkhead transcription factor/forkhead box (FOXO) transcription factors FOXO1/2 to inhibit their function, with important consequences in lymphocytes (27,33) .
4.b. Targets of PI3K: GEFs and Tec.
The control by PI3Ks of the actin cytoskeleton dynamics mediated by GEF stimulation of the Rac and Rho GTPases is well established (21) .One typical GEF is Vav, that is activated by Src and Syk family tyrosine kinases but also depends on its PH and Dbl-homology domains for activity (34) .
With one exception, Tec family tyrosine kinases including BTK and Itk expressed in B and T lymphocytes possess an N terminus PH domain followed by TH, SH3, SH2, and kinase domains (35,36) . The PH domain is determinant to recruitment of these Tec kinases to membranes, where they can further interact with specific Tyr-phosphorylated substrates and activated by Src family and autophosphorylation. Tec family tyrosine kinases have phospholipase C enzymes like PLCg as important substrates within signalosomes generated upon receptor activation. PLCg then splits PtdIns(4,5)P2 into Diacylglicerol and Ins(1,4,5)P3 that in turn are necessary to activate the Ser/Thr kinases PKCq and hence the Ras/MAPK and the IKK/NFkB pathways, or the Ins(1,4,5)P3-Ca2+-dependent activation of NFAT family of transcription factors (Figure 3).
4.c. Targets of PI3K: PDK1-dependent, Akt-independent.
PDK1 can associate to and activate additional substrates that are not dependent on Akt including the Ser/Thr kinases PKCq (37) and the cAMP-dependent kinase PKA (38) , with important functional consequences to lymphocyte activation.
Of note, although the different pathways initiated by PI3K activation have specific targets, they can be also connected to reinforce others. For instance, PKCq-mediated activation of IKK/NFkB is targeted in a PI3-dependent manner through PDK1-, mTORC2- and Tec-dependent mechanisms.
5. Class I-PI3Kinases: Relevance to cancer
The clues for an association of PI3K and tumor development and growth come from different sources. On one hand, some oncogenic retroviruses possess genes derived from those encoding p110a and AKT. On the other, the PtdIns(3,4,5)P3 phosphatase PTEN is a tumor suppressor gene frequently mutated in human tumors with the consequence of a constitutive activation of the PI3K pathway. Another link is the observation of genomic amplification of genes coding for p110a (PIK3CA) or its target Akt (AKT) in several types of cancer (reviewed in (39-41) ).
Although all Class I PI3K subunits have oncogenic potential in vitro; the seminal work by Samuels et al. (42) , later confirmed by abundant data, shows that the p110a catalytic isoform but not other isoforms is mutated with high frequency in colorectal and other different human cancer cells (39-41) ). These gain-of function mutations of the PIK3CA gene are located in the ABD and C2 domains of p110a, but particularly in the helical and catalytic domains. The enzymatic activity is increased by different mechanisms including altered interaction between the ABD and kinase domains of p110a, between the C2 domain and the p85 iSH2 domain, between the p85 nSH2 domain and the helical and kinase domains, or between the kinase domain and the cell membrane, increasing accessibility to phospholipid substrates. Mutations in the PIK3R1 gene coding the p85a regulatory subunit have been also described in different cancer cells. They are clustered in the inter-SH2 domain of p85 that contacts the C2 domain of catalytic subunits or in the nSH2 domain that interacts with the helical domain of catalytic subunits; both might interfere inhibitory interactions between subunits. Interestingly, these cancer-derived gain-of-function mutations in p85a function through the p110a, but not other PI3K catalytic subunits (43) . Last, the p110a isoform of PI3K in endothelium plays a role in tumor growth through its contribution to tumor angiogenesis (44) .
Gain-of-function E1021K mutations of p110d have been recently reported in a residue of the catalytic domain similar to those found in p110a, but these mutations are surprisingly associated with IgM hyperglobulinemia and immunodeficiency rather than cancer (45) . However, p110d can favour tumor growth by several documented mechanisms. Firstly, in some human ovarian and colorectal tumors PIK3CD transcripts can be alternatively spliced to generate a 37 kDa protein (p37δ) that maintains the ABD and RBD domains of p110d. This protein can enhance the proliferation and invasive properties of transformed cells (46) . In second place, p110d activation can indirectly inhibit the activity of the PTEN phosphatase by a mechanism involving RhoA (47) . Enhanced p110d expression in solid tumors in fact suppresses PTEN, contributing to cell growth and –likely- to cancer progression of malignancies of hematopoietic and non-hematopoietic origin (48,49) . The complexity of PI3Kinase activity regulation is further highlighted by the ability of p85a to bind to and to activate PTEN in transformed cell lines (reviewed in (50) ).
6. PI3K inhibitors of broad and restricted specificity. Importance to cancer.
Hence, PI3K inhibitors, or inhibitors of PI3K targets like Akt or mTOR, have been actively looked for as anti-cancer drugs (see (51) , for a recent review). In these assays p110a-, p110b-, p110d-, p110g-, dual p110g/d-specific or pan-PI3K inhibitors, as well as dual PI3K-mTOR inhibitors are under investigation in experimental tests and clinical trials.
7. Role of Class IA and Class IB PI3Kinases in the control of immune responses. Genetic models and PI3K inhibitors
The relevance of class IA PI3K-mediated signals to all steps of innate and adaptive immune responses immediately point to the potential relevance of inhibitors of PI3K or of PI3K effectors as immunomodulators and therapeutic tools in many socially relevant immune-mediated diseases (17,18,22,52-56) . The role of PI3K in immunity has been analyzed in vivo by the generation of mice lacking specific regulatory and catalytic subunits, or expressing kinase-dead mutants of specific subunits, or by means of specific inhibitors of PI3K or its downstream signaling targets. In vitro the analysis has involved cell lines derived from the above models, specific inhibitors, and knock-down approaches using siRNA. We shall now review the genetic and pharmacological data that supports this immunomodulatory potential in the adaptive immune response mediated by B and T lymphocytes.
8. Class IA and IB PI3K in immunity: Lessons from genetic studies in B lymphocytes.
B lymphocytes are an essential arm of the adaptive immune responses as the cells responsible of antibody responses against pathogens; antibodies are produced in great quantities by the B-cell derived plasma cells. The quality of the antibodies both in terms of their affinity for the antigen and the class of the constant regions is essential to efficient removal of parasites, and is largely determined by the cytokines in the milieu, particularly those secreted by antigen-specific T helper cells. Mature B lymphocytes exist in two major subsets, namely the B-1 cells found in body cavities like the peritoneum that have innate properties, and the B-2 cells that comprise non-recirculating MZ (marginal zone) B cells and the FO (follicular) B cells that recirculate through the blood and secondary lymphoid organs.
The role of regulatory subunits has been analyzed in pan-p85a-/- (p85a-/-, p55a-/-, p50a-/-) defective mice, lacking all p85a isoforms. These mice have perinatal lethality, assays using RAG2 chimeric mice with hematopoietic cells derived from mutant ES cells had less mature B cells and low serum immunoglobulins; in vitro activation of B cells was also impaired (57) . Mice lacking p85a subunits but not p55a or p50a had a similar phenotype; both phenotypes were very similar to the X-linked immunodeficiency due to Bruton tyrosine kinase (Btk) deficiency (58) . No immunological defects have been described in mice lacking p55a or p50a subunits (59) . Deletion of p85b produced normal B cell responses (60) . Last, the absence of p85a and/or p85b reduced basal, PI3K dependent lymphocyte motility of B lymphocytes (61) . There is no data concerning the effect of class IB regulatory subunit loss in lymphocyte function, data in neutrophils indicate impaired cytokine-driven motility (62) (summarized in Table 1).
Table 1.- Effect of Class I PI3-K subunit mutations and defects in B and T lymphocyte phenotype and function.
Subunit |
Phenotype, B lymphocyte |
Phenotype, T lymphocyte |
Other (References) |
Class IA regulatory |
|||
Pik3r1 |
|||
pan-p85a-/- (p85a-/-, p55a-/-, p50a-/-) |
Low B cell number; impaired activation and motility |
Impaired motility |
Perinatal lethality (57,61) . |
p85a-/- only |
Impaired motility |
Normal development, reduced motility; enhanced response in vitro; lower T-dependent secondary response |
(60,61,71) .. |
p55a-/-, p50a-/- |
Normal |
Normal |
(59) . |
Pik3r2 |
|||
p85b-/- |
Impaired motility |
Impaired motility |
(61) |
p85a-/-, p55a-/-, p50a-/-; p85b-/-, |
Impaired motility |
Impaired motility, Th2, Treg differentiation; autoimmune syndrome |
(61,72,73) . |
Pik3r3 |
|||
p55g-/- |
n.d. |
n.d. |
|
Class IB regulatory |
|||
Pik3r5 |
|||
p101-/- |
n.d. |
n.d. |
Neutrophil migration impaired (62) |
Pik3r6 |
|||
p87/84 |
n.d. |
n.d. |
Class IA catalytic |
|||
Pik3ca |
|||
p110a CD2Cre (lymphocyte specific deletion) |
Normal development; normal responses in vitro and in vivo |
n.d. |
(63) |
Pik3cb |
|||
p110b CD2Cre (lymphocyte specific deletion) |
Normal |
n.d. |
(63) |
Pik3cd |
|||
p110d-/- |
Impaired B cell homeostasis activation and function |
No alterations detected |
(64,65) |
p110d CD4Cre (T cell specific deletion) |
Impaired Tfh, ICOS signaling |
(74) |
|
p110dD910A/D910A |
Impaired B cell activation and function, enhanced IgE levels |
Impaired T cell activation and function, inflammatory bowel disease |
(66-68) . |
p110a CD2Cre lymphocyte specific deletion; p110dD910A/D910A |
p110d or p110d signal tonic BCR signaling, B cell development and survival; p110d essential in agonist BCR signaling |
n.d. |
(63) |
Class IB catalytic |
|||
Pik3cg |
|||
p110g-/- |
B cells normal |
Altered thymocyte development, impaired or normal activation, impaired migration |
(24,69,75-78) . |
p110g-/- p110d-/- p110g-/- p110dD910A/D910A |
Similar to p110d-/- or p110dD910A/D910A |
Blocked pre-TCR signal, severe T cell high thymocyte apoptosis, severe T cell lymphopenia, Th2-skewed responses, high IgE levels |
(70,79) |
The study of the effect of losing p110a or p110b on lymphocyte development and function has been impaired by the embryonic or early lethality after birth of null (p110a-/- and p110b-/-) or knock-in kinase-dead (p110aD933A and p110bK805R) mutant mice (reviewed in (5) ). Lymphocyte-specific conditional deletion of p110a or p110b in floxed p110a/CD2-Cre or floxed p110b/CD2-Cre mice showed no defect in B lymphocytes (63) . In contrast, p110d-/- (64,65) and p110dD910A mice are viable and have defects in B cell antigen receptor (BCR)-induced activation (63,66) .
In p110d-/- mice Ig in serum and the number of mature B cells were low, B1 and marginal zone B cells were defective, yet T cell numbers and responses were normal. Responses to T-cell independent antigens are low and T-cell dependent responses are severely impaired. B cell antigen receptor (BCR)- and CD40-early signals like calcium flux, activation of phospholipase C, Akt, and Btk, as well as proliferation are impaired (64,65) . These data suggested a specific, unique role for p110d in B cell signaling and function.
The results obtained in mice in which wild type p110d is substituted by the catalytically inactive form p110dD910A were similar concerning B lymphocytes, but in this case T cell antigen receptor (TCR) signaling was also impaired; mice developed inflammatory bowel disease (66) . Furthermore, p110dD910A mice had high serum amounts of antigen-specific IgE antibody despite reduced levels of other isotypes like IgM or IgG1; the same was observed in antigen-specific responses or using p110d-specific inhibitors (67) . This is due to the specific role of p110d-mediated signals in maintaining high levels of the transcription repressor B cell lymphoma 6 (BCL6) acting on the IgE promoter in germinal center (GC) B lymphocytes (68) , where B cells actively proliferate and are induced to produce antibody responses of even higher affinity under the guidance of specialized follicular helper T (Tfh) cells.
Later work by the group of B. Alarcón has shown that, indeed p110d stably binds to BCR and TCR ITAM containing chains through the Ras family protein RRas2 (TC21) actively participating in tonic and ligand activation signaling (15) . Paradoxically, recent data show that patients with a spontaneous dominant gain-of-function mutation of p110d (p110dE1021K) suffer primary immunodeficiency with low IgG2 serum levels, deficient responses to vaccines, lymphopenia and high sensitivity to activation-induced cell death, despite elevated levels of PtdIns(3,4,5)P3 and phosphorylated Akt (45) .
Intriguingly, mice with double lymphocyte deletion of p110a and p110dD910A indicates that p110a could substitute for p110d in agonist-independent tonic BCR signaling necessary for B cell development from B cell progenitors and B cell survival; however p110a could not substitute for p110d in agonist BCR activation (63) .
Both p110d and p110g catalytic subunits are highly expressed in lymphocytes but not in other cell types that are not of hematopoietic origin. However, mice p110g-/- had no B cell defects but show altered differentiation in the thymus, with decreased CD4+ numbers, impaired migration and survival of mature thymocyte as well as mature T cell activation, (69) . Mice deficient in both p110g and p110d (p110g-/- p110dD910A) have a B cell phenotype similar to that of their p110g-sufficient counterparts (70) .
9. Class IA and IB in immunity: Lessons from genetic studies: T lymphocytes.
In mice with hematopoietic cells lacking all p85a-/-, p55a-/-, p50a-/- subunits the number and in vitro activation of T cells was largely normal (57) ; mice lacking p85a subunits but not p55a or p50a had a similar phenotype (58) . No immunological defects have been described in mice lacking p55a or p50a subunits (59) . Deletion of p85b induced enhanced T-lymphocyte responses (60) , however loss of p85b impaired secondary T cell dependent antigen responses by CD28-mediated mechanisms (71) . Conditional deletion of the class IA regulatory subunits p85a, p55a, p50a, and p85b in the T cell lineage (p55g is undetectable in T lymphocytes, J.M.R., unpublished data) resulted in normal T cell development and Th1 differentiation. PI3K signals, antibody responses to T-cell dependent antigens, Th2, and Treg differentiation were impaired and the animals could develop an autoimmune Sjögren’s-like syndrome (72,73) . As in B lymphocytes, loss of p85a and p85b reduced basal lymphocyte motility (61) .
In p110d-/- mice the number of quality of T lymphocytes appear normal; and decreased responses to T-cell dependent antibody responses might be largely due to defects in the B cell compartment (64,65) . Mice where wild type p110d is substituted by the catalytically inactive form p110dD910A were similar concerning B lymphocytes, but in this case T cell antigen receptor (TCR) signaling was also impaired; mice developed inflammatory bowel disease (66) . Later work has shown that the p110δ subunit was key to ICOS-mediated costimulation of effector function in follicular helper T cells (74)
Mice with deleted p110g subunits show altered differentiation in the thymus, with decreased CD4+ numbers, impaired migration and survival of mature thymocyte as well as mature T cell activation (69,75) possibly because p110g binds to and is activated by TCR ligation (24) . However, other groups have found no defects in CD4+ T cell signaling and proliferation but did show strong defects in migration and trafficking in TCR transgenic mice (76,77) ; Martin et al. (78) observed no effect in the migration and trafficking of naive CD8+ T cells, or in their proliferation and activation migration. In contrast, migration of p110g-deficient CD8+ effector T cells induced by inflammatory stimuli was impaired (78) .
Double knockout mice (p110g-/-p110d-/-) deficient for both p110g and p110d have severe defects in T cell differentiation as thymocytes are blocked in the DN3-DN4 selection checkpoint of pre-TCR signaling, with low thymocyte numbers and high apoptosis (79) . Mice lacking p110g and expressing the inactive mutant p110dD910A (p110g-/- p110dD910A) have similarly strong defects in thymus differentiation, but the few remaining T cells infiltrate the mucosas, show Th2-skewed responses and anomalously high IgE levels (70) .
What is perhaps more surprising of the results using genetic models, is the certain selectivity of PI3K subunits in terms of the lymphocyte population(s) and processes affected. This applies not only to PI3K subunits, but also to other protein targets of PI3K, giving clues as to the possibility of selective immune intervention using PI3K inhibitors or its downstream effector molecules in autoimmune diseases or other immune-based illnesses. The importance of PI3K in different types of cancer has boosted a very active search for small pharmacological inhibitors directed at one or several PI3K subunits and/or PI3K downstream targets like Akt or mTOR. These inhibitors have the potential of being used to modulate immune responses. The use of inhibitors, though, has to take into account the harming potential to the host, particularly in long-term treatment of chronic diseases. These adverse effects can be due to off-target effects of a particular drug, but also to on-target effects of inhibitors of widely distributed enzymes fulfilling essential functions, as are PI3Ks. However, pre-clinical models and clinical trials in cancer indicate that treatments with PI3K inhibitors can be well tolerated, even when using inhibitors of the widely expressed p110a and p110b, or even pan-class I PI3K inhibitors (51) . Another aspect to be taken into account is that, like any other immunosuppressive drug, PI3K inhibitors are likely to have undesired side effects including increased susceptibility to infection (4) .
10. Class IA and IB PI3K in immunity: Lessons from specific inhibitors in B lymphocyte function
As mentioned before, genetic defects in p110d affect particularly B-1 and B-2 marginal zone B lymphocytes; the similarity of these defects and those of mice lacking Akt or the PI3K-recruiting costimulatory molecule CD19 might indicate a role of a CD19/p110d PI3K/ Akt/ Foxo1 downstream signal in B-1 and MZ B-2 differentiation (reviewed in (22) ). Using the p110d inhibitor IC87114, Bilancio et al. (80) found strong inhibition of anti-BCR-induced proliferation of B lymphocytes, Ca2+ flux, or phosphorylation of downstream targets like Akt, or the Akt targets forkhead transcription factor/forkhead box O3a (FOXO3a), and p70 S6 kinase (p70 S6K), and partial inhibition of extracellular signal-regulated kinase (Erk), that mimicked p110d defects. In contrast, the p110g/a inhibitor AS-604850 had no detectable effect on these parameters (80) . Despite the fact that p110d, rather than p110a catalytic subunits of PI3K, had detectable effects in B lymphocytes and B cell responses, and p110d binds to the BCR (15) , So et al. recently analyzed highly specific p110a inhibitors to assess a possible role for the p110a-subunit in B cell function (81) . They observed a minor but clear effect of p110a-specific inhibitors A66 and MLN1117 on early Akt activation and Ca2+ flux, or B cell proliferation induced by BCR stimuli in vitro, but not by cytokines IL-4 or BAFF. This inhibition could add to the stronger inhibition induced by the p110d inhibitor IC87114 on the same phenomena, that was not complete (81) .
In vivo, unlike administration of MLN1117, daily administration of a p110d (IC87114) or a pan-PI3K inhibitor (GDC-0941) to immunized mice strongly reduced marginal zone B cell numbers in the spleen; GDC-0941 also reduced the number of germinal centers (81) . Despite these effects, antigen specific IgM, IgG1 titers in the serum of immunized mice was not significantly lower or were elevated, and in fact IgE titers were significantly enhanced by the p110d inhibitor IC87114 or the pan-PI3K inhibitor GDC-0941, but not by p110a inhibitors (81) . Our own results with a dual p110a/d inhibitor (ETP-46321) administered after initiation of antigen responses showed inhibition of antigen-specific serum IgG3, but not of IgM, IgG1, or IgG2b antibody (82) .
As mentioned above, p110a inhibition did not affect B cell Akt and FOXO signaling or survival induced by cytokines like IL-4 or BAFF, yet p110d inhibitors, pan-PI3K inhibitors, and to a lesser extent, also p110b inhibitors like TGX-221 produced significant results (80,81) .
It has been observed that p110d signals play an essential role in inhibiting excess IgE production by maintaining the levels of the BCL6 that negatively controls the IgG1 to IgE switch. These effects can be mimicked in vitro or in vivo using p110d-specific inhibitors like IC87114, or broad-spectrum PI3K inhibitors like PIK-90 and PI-103, or GDC-0941, but not by p110a inhibitors like MLN1117 (67,68,81) .
11. Class IA and IB PI3K in immunity: Lessons from specific inhibitors in T lymphocytes
Early data by Shi et al. using the PI3K inhibitor Wortmannin indicated that the role of PI3K in (CD4+) T lymphocyte activation was dependent on the stimulus used. Production of IL-2 by the surrogate antigen activation anti-CD3 antibodies plus CD28 costimulus seemed largely PI3K-independent, whereas activation and Th2 differentiation of the same cells by peptide antigen and antigen-presenting cells was clearly inhibited by Wortmannin (83) . Although the specificity of Wortmannin for PI3K has been questioned (see, i.e. (9,84) ), later studies by Okkenhaugh et al. in mice expressing the inactive p110dD910A mutant also found that antigen activation, rather that activation by anti-CD3 plus anti-CD28, was impaired in the mutant mice (66,85) . Furthermore, differentiation of p110dD910A CD4+ T lymphocyte into the Th1 (IFN-g-producing) and Th2 (IL-4-producing) T helper cell subsets was also impaired. The p110d inhibitor IC87114 inhibited Th1 differentiation, but Th17 differentiation was even more sensitive to inhibition (86) . In contrast, we found that in cultures of naive CD4+ T lymphocytes activated with anti-CD3 plus anti-CD28 antibodies under neutral conditions IFN-g-production was highly sensitive to inhibition of PI3K, at doses ten-fold lower than those needed to inhibit proliferation, IL-2, or IL-10. The p110d inhibitor IC87114 or the dual p110a/d inhibitor ETP-46321, were particularly effective, whereas inhibition by A66, a p110a inhibitor was not as marked (82) . A differentially high inhibition of IC87114 on IFN-g production versus proliferation was also observed by Okkenhaugh’s group (87) . Furthermore, these authors observed that isolated memory/effector cells were more sensitive to the p110d inhibitor IC87114 that naive cells. These results paved the way to show inhibition by IC87114 of anti-CD3 antibody-mediated T cell activation or recall responses to vaccines in human cells (87) . In our experience, though, inhibition of cytokine production by CD4+ blast cells activation with anti-CD3 or anti-CD3 plus anti-ICOS antibodies was similar to that observed in activation of naive CD4+ T cells (82) .
In contrast, So et al. observed no significant effect of p110a inhibition by A66 or MLN1117 on antigen- or mitogen- induced CD4+ T cell proliferation, or secretion of IL-2 and IFN-g, although there was some inhibition of Akt phosphorylation, and p110a inhibitors enhanced the clear but not complete inhibition induced by IC87114 (81) . A contribution of p110a to TCR and costimulation signaling is also suggested in the response of the CD4+ T cell line D10 to anti-CD3 and anti-ICOS (14) . In this system, we observed partial inhibition of early Akt phosphorylation by the p110a inhibitor PIK-75 or by p110a silencing (14) . Intriguingly, whereas silencing p110d or inhibition with IC87114 clearly inhibited both Akt and Erk activation, p110a silencing markedly enhanced Erk phosphorylation, suggesting that p110a might exert a negative control over some p110d signals (14) .
Follicular helper T cells (Tfh) are particularly and functionally very important to the development germinal centers and of efficient antibody responses, promoting antibody affinity maturation and differentiation of memory cells. They characteristically express the surface molecules CXCR5, PD-1 and ICOS, as well as the transcription suppressor Bcl6 (88) . Although Tfh cells able of producing different antibody-promoting cytokines (IL-4, IL-10, IL-17, or IFN-g) can be found in vivo, IL-21 is the most characteristic cytokine of Tfh cells. Signaling by the PI3K-binding costimulators CD28 and ICOS are needed for efficient development of Tfh and germinal centers. Work with mice expressing ICOS mutants unable to bind PI3K, or in mice expressing the inactive p110dD910A kinase, or inhibition with the p110d inhibitor IC87114, taken together indicate that PI3K p110d is needed for Tfh differentiation and antibody production in a ICOS dependent way; early TCR signaling or IL-21 production in differentiated Tfh is also potentiated by ICOS signaling and inhibited by IC87114 (74,89) . So et al. have recently described that IC87114 but not the p110a inhibitor MLN1117 inhibits germinal center formation (81) . Our own data indicates that ICOS enhances early TCR activating signals and IL-17A production in Tfh in a p110d-dependent fashion, yet IL-21 secretion is largely independent of ICOS and inhibition of both p110d and p110a is needed to block IL-21 production (82) .
ICOS and its PI3K associated activity intervene in yet another aspect of germinal center development, namely in the recruitment of activated T helper cells into the follicle. ICOS is directly involved in this movement, but this effect is independent on costimulation, yet is dictated by the interaction of ICOS in T cells and its ligand ICOS-L expressed in bystander B lymphocytes in a PI3K-dependent fashion (90) . ICOS ligation, as ligation of other CD28 family members (CD28, CTLA-4/CD152) can induce antigen-independent, tyrosine kinase and PI3K-dependent changes in actin cytoskeleton and cell elongation and spreading (14,91-95) . However, Xu et al. found that recruitment of T cells into follicles was dependent on the p110d PI3K subunit (90) , whereas using a Th2 T helper cell line we have found that cell elongation was dependent on p110a, rather than p110d (14) .
Another functionally important T cells are the CD4+ regulatory T (Treg) cells that express the transcription factor Foxp3 and negatively control adaptive immune responses, as they expand during normal antigen responses to help in their termination. They can differentiate from immature precursors in the thymus (tTreg cells), but also from mature CD4+ lymphocytes in the periphery (pTreg cells). A role for p110d in Treg function is suggested by data from mice transgenic for the kinase-dead p110dD910A mutation. These mice have reduced numbers of Treg cells with reduced suppressor capability in vitro and in vivo (96) . Paradoxically, this deficit in Treg function can help in producing efficient anti-parasite responses in certain cases. Unlike wild type mice, animals lacking p110d can efficiently clear Leishmania infection because of strong defects in Treg number and homing (97) . It is also paradoxical that the proportion of Treg differentiated in vitro (iTreg) can be fostered by inhibitors of PI3K or mTOR, yet this is mainly due to p110a, as judged by the effect of inhibitors of p110a, p110b or p110d (98,99) . In contrast, IL-2 dependent PI3K signals are necessary for Treg expansion, and high PI3K signals due to PTEN deficiency do not affect Treg function in vitro or in vivo (100) .
The last T cell subset to be considered is the cytotoxic, MHC class I-restricted CD8+ population of T lymphocytes that has a prime role in the response against intracellular pathogens like viruses as well as in anti-tumor responses. There are conflicting data on the role of PI3K in TCR-mediated activation in CD8+ T cells. According to Ni et al., proliferation and induction of cytotoxic activity induced by TCR/CD3 and CD28 ligands was not affected by the PI3K inhibitor Wortmannin (101) . In contrast, Phu et al. observed clear inhibition of anti-CD3-induced proliferation and cytotoxic function in CD8+ T lymphocytes cultured in the presence of Wortmannin or LY294002 (102) . More recently, Soond et al. found a clear inhibition by LY294002 or the p110d inhibitor IC87114 on antigen-induced proliferation or IL-2 production in CD8+ T lymphocytes; inhibition was complete when IFN-g was analyzed. D. Cantrell’s group has recently analyzed the role of PI3K in IL-2-induced expansion of previously activated CD8+ T lymphocytes. Interestingly, although IL-2 activated PI3K-dependent activation of Akt phosphorylation in these cells, inhibitors of PI3K like IC87114 blocked IL-2-dependent Akt phosphorylation but not CD8+ proliferation, that was dependent on PDK1 (103) . Still, blocking p110d or Akt enhanced the expression of molecules involved in lymph node homing and homing itself, showing their immunomodulatory potential in these cells.
12. PI3K in pathological immune responses
Given the relevant role of PI3K in immune responses, particularly the p110g and p110d of high expression in leukocytes, there has been an obvious interest in analyzing the possible use of PI3K inhibitors in both organ-specific and systemic autoimmune diseases of social impact including rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, or its animal experimental models. Although we shall focus on the role of adaptive immune response in these diseases, it is important to remember that PI3K have an essential role in the function of cells of the innate arm of immunity like macrophages and dendritic cells, polymorphonuclear leukocytes, mast cells, or natural killer (NK) lymphocytes (52) . So, the ultimate effect of PI3K inhibitors in immune responses in vivo will largely depend on their joint effects on the development, expansion, mobility, and activation of effector function in innate and adaptive immunity elements.
12.a. Rheumatoid arthritis.
Rheumatoid arthritis (RA) affects a significant fraction of the population. It is an immune-mediated, chronic inflammatory disease of the joints that are infiltrated by leukocytes. This eventually leads to the formation of pannus, the damage of cartilage and bone erosion. From many data, including those from animal experimental models it is assumed that in the development of arthritis participate immune response cellular elements from both the innate (macrophages, neutrophils, mast cells) and adaptive (T and B lympocytes) immunity.
Data from mice lacking p110g catalytic subunits indicate that these mice are resistant to the development of clinical symptoms of arthritis. Oral administration of the p110g inhibitor AS-605240 ameliorated the symptoms in two experimental models of arthritis, the collagen-induced arthritis (CIA) dependent on T-cell dependent cytokines and B cell help, and in the neutrophil-dependent arthritis induced by anti-collagen II monoclonal antibodies (104) . Similarly, in a neutrophil-dependent model of arthritis induced by serum from arthritis K/BxN mice, clinical symptoms were dependent on p110d and p110g, and IC87114 inhibited progression of the disease (105) . In the rat model of adjuvant-induced arthritis (AIA), therapeutic oral administration of the wide spectrum phosphatidylinositol 3-kinase inhibitor ZSTK474 ameliorated clinical signs of the disease and inhibited the production of the effector cytokines IFN-g and IL-17 by T cells in vitro, or the proliferation and prostaglandin E2 (PGE2) production by fibroblast-like synoviocytes cells (FLS) (106) . PI3Kg deficiency in a model of arthritis induced by transgenic expression of human TNF reduced metalloproteinase secretion by fibroblasts, cartilage damage and clinical score, yet did not affect recruitment of inflammatory leukocytes (107) . The p110g-specific inhibitor AS252424 also inhibited invasiveness and matrix metalloproteinase secretion by human synovial fibroblasts from patients with RA. Intriguingly, in FLS from rheumatoid arthritis patiens PI3K p110d was specifically induced in FLS from rheumatoid arthritis patients by pro-inflammatory cytokines like tumor necrosis factor (TNF), TNF signaling as well as platelet-derived growth factor (PDGF)-dependent synoviocyte growth were inhibited by PI3K p110d inhibitors CAL-101 or INK007, but not by p110a inhibitors like A66 (108) .
12.b. Systemic Lupus Erythematosus.
Systemic Lupus Erythematosus (SLE) is an autoimmune, chronic, inflammatory, multisystemic disease with a particularly high incidence in females. SLE is due to tissue and cell damage induced by autoantibodies generated by early expansion of long-lived autoreactive T helper memory cells that trigger polyclonal hyperactivity of B lymphocyte responses and hypergammaglobulinemia; expansion of autoreactive B cells leads to enhanced levels of antibodies against a variety of self antigens, particularly nuclear antigens and DNA. Accumulation of immune complexes in the kidney leads to inflammatory reaction ultimately leading to glomerulonephritis.
There are different animal models that spontaneously develop a SLE-syndrome, including the widely used MRL-lpr and MRL-gld mice with a mutation in the expression of the apoptosis-signaling Fas molecule or its ligand, respectively. Roquinsan/san mice have a mutation that produce disregulated expression of the PI3K binding costimulatory molecule ICOS, show progressively enhanced numbers of Tfh, of germinal centers and autoantibodies, and nephritis (109,110) with a prime role for Tfh IFN-g (111,112) .
The effect of PI3K inhibitors on lupus has been analyzed in different models. In the MRL-lpr model of lupus, Barber et al. described a preventive and therapeutic effect of the p110g inhibitor AS605240 on autoantibodies, proteinuria, and reduced number of pathogenic CD4+ memory T lymphocytes due to enhanced cell death (113) , a finding that was also observed in the p65PI3K transgene mouse model of lupus when p110g was deleted (114) . Resistance to activation-induced cell death in T lymphocytes is an important factor to SLE development, as shown in the enhanced PI3K activity in lymphocytes from SLE patients due to higher p110d (115) ; this enhanced resistance to activation induced cell death was abolished, upon inhibition of p110d with IC87114, particularly in the memory T cells that are enhanced in these patients (115) . Yet another model of SLE is the Lyn deficient mice. Lyn-/- mice have enhanced PI3K signaling, and simple attenuation of p110d signals in haploinsuficient Lyn−/−110δ+/ D910A mice ameliorates the disease, with lower autoantibody levels and nephritis by a mechanism that involved lower T cell activation rather than attenuation of B lymphocyte responses (116) .
12.c. Multiple sclerosis (MS) and Experimental Autoimmune Encephalomyelitis (EAE).
Multiple sclerosis is an inflammatory disease characterized by demyelination of the central nervous system (CNS). Experimental autoimmune encephalomyelitis is the model of choice for MS in mice, and is induced by immunization with proteins or protein fragments in susceptible strains of mice. Inflammation and axonal damage is si accompanied by infiltration of T lymphocytes and other leukocytes, and in certain cases by the formation of structures resembling lymphoid follicles; these contain T and B cells, plus professional antigen presenting cells.
T lymphocytes specific for myelin antigens that secrete IFN-g, IL-17, or IL-9 are able of inducing EAE, hence it is reasonable to assume that therapeutic approaches that suppress the secretion of these cytokines can be useful in controlling the evolution of the disease. Mice expressing the kinase-dead mutant 110δD910A had a milder form of EAE induced by the rat Myelin Oligodendrocyte Glycoprotein (MOG) peptide35-55, and lower number of lesions and T cell inflammatory infiltration, plus enhanced apoptosis (86) . As discussed in a previous section, in this study the p110d inhibitor IC87114 inhibited the differentiation of IFN-g producing Th1 cells and IL-17 producing Th17 cells, yet Th17 more effectively than Th1, indicating a potential for PI3K as a target in MS therapy. We have recently used an oral therapeutic treatment with the dual p110a and DNA-PK inhibitor PIK-75 to significantly inhibit MOG-induced EAE symptoms (117) . This was accompanied by in vitro observations showing that PIK-75 induced cell death in resting or activated T and B lymphocytes, or CD4+ T lymphocyte proliferation and cytokine (IL-2, IFN-g, IL-17A, or IL-21) secretion in the nanomolar range. Intriguingly, we have later found that other, more specific p110a inhibitors like A66 do not have a similar effect on lymphocyte apoptosis (82) , and it is probably the sum of p110a and DNA-PK inhibition that produced the effect on cell viability (unpublished data).
12.d. Experimental colitis and psoriasis.
Oral treatment with PIK-75 was previously shown to inhibit another experimental inflammation, namely colitis induced by oral administration of dextran-sulfate, a model for human inflammatory bowel disease (118) . In vitro, PIK-75 inhibited AKT phosphorylation, IKK activation, and NF-kB transcription in lymphoid or monocyte cells or cell lines, secretion of the inflammatory cytokines IL-6 and TNF-a by activated monocytes, or reduced the expression of adhesion molecules by TNF-a in endothelial cells (118) . As mentioned before, functional CD4+ Treg cells are essential to prevent the development of experimental colitis, and mice expressing the kinase-dead mutant 110δD910A cannot prevent the development of the disease, suggesting the importance of the p110d isoform to the effector function of Treg cells (96)
Last, PI3K have a role in the psoriasis-like dermatitis induced by imiquimod (IMQ). In this model, the T lymphocytes of the gd TCR subset produce IL-17 essential to the clinical symptoms of the disease. After establishing that mice transgenic for the 110δD910A inactive mutant or p110g-/- knockouts are not susceptible to dermatitis and have diminished production of IL-17A and F (119) , Roller et al. went on to show that the p110d inhibitor IC87114 or p110g inhibitors like AS605240 inhibited IL-17 and IFN-g production by activated peripheral blood lymphocytes from psoriatic and healthy donors, or IFN-g production by activated blood TCRγδ T lymphocytes (119) .
13. PI3K and cancer: Impact of PI3K inhibitors on cancer cells and anti-cancer immunity: A double-edged sword?
Tumor cells can express tumor antigens of various kinds that can elicit efficient immunity as an extrinsic mechanism to control and suppress cancer growth (immunosurveillance), establishing a dynamic equilibrium or even complete rejection of tumor cells. However, tumor specific responses can also promote tumor growth through the selection of rare mutant tumor cells able of oppose or evade the immune mechanisms developed by the host (immunoediting) (120,121) . CD8 T lymphocytes and IFN-g seem to be major cellular and molecular mediators of anti-cancer responses. One mechanism used by tumor cells to evade immune-mediated rejection is by expressing inhibitory B7 family molecules like B7-H1 and B7-H4 that bind the inhibitory CD28 family ligand programmed death-1 (PD-1) expressed on the surface of activated T cells, B cells and macrophages (122) . A B7 family receptor expressed by melanoma and other tumor cells is the ICOS ligand that can directly induce the activation and expansion of ICOS-expressing Treg cells within the tumor, suppressing anti-tumor rejection, or alternatively induce the recruitment to the tumor of ICOS-ligand-expressing dendritic cells that favor Treg function (123-127) . Another inhibitory CD28-like molecule expressed by Treg cells is CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, CD152). It is not surprising, then, that antibodies blocking CTLA-4 or the PD-1/PD1 ligands are currently used in anti-cancer therapy, supposedly to unleash suppressed anti-tumor immunity (128) .
Since p110a are frequently mutated in cancer, PI3K inhibitors directed at this isoform, alone or together with other kinases, are currently being tested in antitumour therapies (51) . Although in our experience inhibition of p110a, alone or together with DNA-PK can have a significant impact in immune responses and lymphocyte viability (82,117) , other data suggest minimal damage of p110a inhibition to immune reactions and likely to anti-tumor immunity (81) . Thus, it might seem that only broad PI3K inhibitors, or Akt and mTOR inhibitors, might have a significant impact on anti-tumor responses, whereas immunosuppressive p110d- or p110g-specific inhibitors would be of no interest to cancer therapy, except in hematologic malignancies were the levels of these subunits is naturally high. Interestingly, p110d participates in antigen, cytokine and chemokine receptor signaling in B lymphocytes, and indeed one p110d inhibitor CAL-101 has been used in the treatment of some relapsed or refractory B-cell malignancies including chronic lymphocitic leukemia (CLL), non-Hodgkin’s lymphoma (NHL), acute myeloid leukemia (AML), and multiple myeloma (MM), with clear clinical responses in CLL and some NHL (49,129) . Surprisingly, this is due in part to inhibition of signals delivered by the tumor environment that sustain leukemia and lymphoma cells.
It is yet more surprising the data showing that p110d inhibitors can be useful in the treatment of solid tumors that do not harbor p110d mutations. Tzenaki et al. have reported that breast and prostate cancer cells can possess high levels of p110δ that not only enhance cellular PI3K activity but also indirectly inhibit the tumor suppressor phosphatase PTEN via RhoA and ROCK activation, further enhancing PtdIns(3,4,5)P3 levels (48,49) . Thus, p110δ-selective PI3K inhibitors might be useful in certain solid tumor therapy by directly inhibiting p110δ signaling as well as by indirectly activate PTEN to diminish general PtdIns(3,4,5)P3 levels. At the same time, the use of p110d inhibitors in these cases does have a true danger for suppressing anti-tumor immune responses, but also to responses against pathogens.
14. Concluding remarks
Research in recent years has accumulated evidence showing the essential role of class I PI3K and their molecular targets in cancer and immune responses, and the potential benefits of PI3K inhibitors to treat neoplastic and autoimmune diseases. The therapeutic potential of PI3K inhibitors in cancer has prompted the discovery of many different molecules by the pharmaceutical industry whose utility in immune-based diseases needs to be tested.
Many of the newly developed PI3K inhibitors also inhibit other molecules like mTOR or DNA-PK fulfilling important functions in different types of cells, so that chronic treatment with these drugs might produce deleterious effects on the host in the long term (129) . According to currently available data, the PI3K inhibitors under clinical or preclinical study are reasonably well tolerated, and these include broad specificity PI3K inhibitors or dual PI3K and mTOR inhibitors (51) . The challenge now is to determine which particular type(s) of inhibitor(s) are of real benefit to each particular disease.
Acknowledgements
P.P. is a Tenured Scientist of the Consejo Superior de Investigaciones Científicas (CSIC) at the Centro Nacional de Microbiología, Instituto de Salud Carlos III. Supported by Grants PI10/00650 and PI13/02153 (to JMR) and PI10/00648 and PI13/01809 (to PP) from “Acción Estratégica en Salud, Plan Estatal I+D+i”, Ministerio de Economía y Competitividad (MINECO), Spain. The authors apologize to the many authors whose relevant data have been summarized or omitted due to space constraints.
Disclosures
The authors declare no financial conflict of interest.
references
1. Vanhaesebroeck, B.; Leevers, S. J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P. C.; Woscholski, R.; & al. Synthesis and function of 3-phosphorylated Inositol lipids. Annu Rev Biochem 70, 535-602 (2001).
2. Deane, J. A.; & Fruman, D. A. Phosphoinositide 3-Kinase: Diverse roles in immune cell activation. Annu Rev Immunol 22, 563-598 (2004).
3. Hawkins, P. T.; Anderson, K. E.; Davidson, K.; & Stephens, L. R. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans 34, 647-662 (2006).
4. Fruman, D., A.; & Bismuth, G. Fine tuning the immune response with PI3K. Immunol Rev 228, 253-272 (2009).
5. Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; & Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 11, 329-341 (2010).
6. Lempiainen, H.; & Halazonetis, T. D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J 28, 3067-3073 (2009).
7. Lees-Miller, S. P. PIKK-ing a new partner: A new role for PKB in the DNA damage response. Cancer Cell 13, 379-380 (2008).
8. van der Burg, M.; Ijspeert, H.; Verkaik, N. S.; Turul, T.; Wiegant, W. W.; Morotomi-Yano, K.; Mari, P.-O.; & al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest 119, 91-98 (2009).
9. Knight, Z. A.; Gonzalez, B.; Feldman, M. E.; Zunder, E. R.; Goldenberg, D. D.; Williams, O.; Loewith, R.; & al. A pharmacological map of the PI3-K family defines a role for p110a in Insulin signaling. Cell 125, 733-747 (2007).
10. Kurig, B.; Shymanets, A.; Bohnacker, T.; Prajwal; Brock, C.; Ahmadian, M. R.; Schaefer, M.; & al. Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110g. Proc Natl Acad Sci USA 106, 20312-20317 (2009).
11. Huang, C.-H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V. E.; Kinzler, K. W.; Vogelstein, B.; & al. The structure of a human p110a/p85a complex elucidates the effects of oncogenic PI3Ka mutations. Science 318, 1744-1748 (2007).
12. Backer, J. M. The regulation of Class IA PI 3-Kinases by inter-subunit interactions. In Rommel, C.; Vanhaesebroeck, B.; & Vogt, P. K., Editors. In Phosphoinositide 3-kinase in Health and Disease; Springer Berlin Heidelberg, 2010; p. 87-114.
13. Fruman, D. A. Regulatory Subunits of Class IA PI3K. In Rommel, C.; Vanhaesebroeck, B.; & Vogt, P. K., Editors. In Phosphoinositide 3-kinase in Health and Disease; Springer Berlin Heidelberg, 2010; p. 225-244.
14. Acosta, Y.; Zafra, M.; Ojeda, G.; Bernardone, I.; Dianzani, U.; Portolés, P.; & Rojo, J. Biased binding of class IA phosphatidyl inositol 3-kinase subunits to inducible costimulator (CD278). Cell Mol Life Sci 68, 3065-3079 (2011).
15. Delgado, P.; Cubelos, B.; Calleja, E.; Martinez-Martin, N.; Cipres, A.; Merida, I.; Bellas, C.; & al. Essential function for the GTPase TC21 in homeostatic antigen receptor signaling. Nat Immunol 10, 880-888 (2009).
16. Rodriguez-Viciana, P.; Sabatier, C.; & McCormick, F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol 24, 4943-4954 (2004).
17. Parry, R. V.; Riley, J. L.; & Ward, S. G. Signalling to suit function: tailoring phosphoinositide 3-kinase during T-cell activation. Trends Immunol 28, 161-168 (2007).
18. Huang, Y. H.; & Sauer, K. Lipid signaling in T-cell development and function. Cold Spring Harb Perspect Biol 2, a002428 (2010).
19. Ward, S. G.; & Blunt, M. D. Pharmacological targeting of phosphoinositide lipid kinases and phosphatases in the immune system: Success, disappointment and new opportunities. Front Immunol 3, 226 (2012).
20. Fos, C.; Salles, A.; Lang, V.; Carrette, F.; Audebert, S.; Pastor, S.; Ghiotto, M.; & al. ICOS ligation recruits the p50a PI3K regulatory subunit to the Immunological Synapse. J Immunol 181, 1969-1977 (2008).
21. Ward, S. G.; & Cantrell, D. A. Phosphoinositide 3-kinases in T lymphocyte activation. Curr Opinion Immunol 13, 332-338 (2001).
22. So, L.; & Fruman, D. A. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J 442, 465-481 (2012).
23. Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; & al. The p110b isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110g. Proc Natl Acad Sci USA 105, 8292-8297 (2008).
24. Alcazar, I.; Marques, M.; Kumar, A.; Hirsch, E.; Wymann, M.; Carrera, A. C.; & Barber, D. F. Phosphoinositide 3 kinaseg participates in T cell receptor induced T cell activation. J Exp Med 204, 2977-2987 (2007).
25. Nombela-Arrieta, C.; Lacalle, R. A.; Montoya, M. C.; Kunisaki, Y.; Megías, D.; Marqués, M.; Carrera, A. C.; & al. Differential requirements for DOCK2 and Phosphoinositide-3-Kinase g during T and B lymphocyte homing. Immunity 21, 429-441 (2004).
26. Reif, K.; Okkenhaug, K.; Sasaki, T.; Penninger, J. M.; Vanhaesebroeck, B.; & Cyster, J. G. Cutting edge: Differential roles for Phosphoinositide 3-Kinases, p110g and p110d, in lymphocyte chemotaxis and homing. J Immunol 173, 2236-2240 (2004).
27. Manning, B. D.; & Cantley, L. C. AKT/PKB signaling: Navigating downstream. Cell 129, 1261-1274 (2007).
28. Yap, T. A.; Garrett, M. D.; Walton, M. I.; Raynaud, F.; de Bono, J. S.; & Workman, P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol 8, 393-412 (2008).
29. Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 12, 325-338 (2012).
30. Dodson, L. F.; Boomer, J. S.; Deppong, C. M.; Shah, D. D.; Sim, J.; Bricker, T. L.; Russell, J. H.; & al. Targeted kock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol Cell Biol 29, 3710-3721 (2009).
31. Dragoi, A.-M.; Fu, X.; Ivanov, S.; Zhang, P.; Sheng, L.; Wu, D.; Li, G. C.; & al. DNA-PKcs, but not TLR9, is required for activation of Akt by CpG-DNA. EMBO J 24, 779-789 (2005).
32. Bozulic, L.; Surucu, B.; Hynx, D.; & Hemmings, B. A. PKBa/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 30, 203-213 (2008).
33. Finlay, D.; & Cantrell, D. A. Phosphoinositide 3-kinase and the mammalian target of rapamycin pathways control T cell migration. Ann N Y Acad Sci 1183, 149-157 (2010).
34. Bustelo, X. R. Regulatory and signaling properties of the Vav family. Mol Cell Biol 20, 1461-1477 (2000).
35. Smith, C. I. E.; Islam, T. C.; Mattsson, P. T.; Mohamed, A. J.; Nore, B. F.; & Vihinen, M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. BioEssays 23, 436-446 (2001).
36. Readinger, J. A.; Mueller, K. L.; Venegas, A. M.; Horai, R.; & Schwartzberg, P. L. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol Rev 228, 93-114 (2009).
37. Lee, K.-Y.; D'Acquisto, F.; Hayden, M. S.; Shim, J.-H.; & Ghosh, S. PDK1 nucleates T cell receptor-induced signaling complex for NF-kB activation. Science 308, 114-118 (2005).
38. Nirula, A.; Ho, M.; Phee, H.; Roose, J.; & Weiss, A. Phosphoinositide-dependent kinase 1 targets protein kinase A in a pathway that regulates interleukin 4. J Exp Med 203, 1733-1744 (2006).
39. Zhao, L.; & Vogt, P. K. Class I PI3K in oncogenic cellular transformation. Oncogene 27, 5486-5496 (2008).
40. Samuels, Y.; & Waldman, T. Oncogenic mutations of PIK3CA in human cancers. In Rommel, C.; Vanhaesebroeck, B.; & Vogt, P. K., Editors. In Phosphoinositide 3-kinase in Health and Disease; Springer Berlin Heidelberg, 2010; p. 21-41.
41. Gabelli, S.; Huang, C.-H.; Mandelker, D.; Schmidt-Kittler, O.; Vogelstein, B.; & Amzel, L. M. Structural effects of oncogenic PI3Ka mutations. In Rommel, C.; Vanhaesebroeck, B.; & Vogt, P. K., Editors. In Phosphoinositide 3-kinase in Health and Disease; Springer Berlin Heidelberg, 2010; p. 43-53.
42. Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; & al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004).
43. Sun, M.; Hillmann, P.; Hofmann, B. T.; Hart, J. R.; & Vogt, P. K. Cancer-derived mutations in the regulatory subunit p85a of phosphoinositide 3-kinase function through the catalytic subunit p110a Proc Natl Acad Sci USA 107, 15547-15552 (2010).
44. Soler, A.; Serra, H.; Pearce, W.; Angulo, A.; Guillermet-Guibert, J.; Friedman, L. S.; Viñals, F.; & al. Inhibition of the p110a isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J Exp Med 210, 1937-1945 (2013).
45. Angulo, I.; Vadas, O.; Garçon, F.; Banham-Hall, E.; Plagnol, V.; Leahy, T. R.; Baxendale, H.; & al. Phosphoinositide 3-kinase d gene mutation predisposes to respiratory infection and airway damage. Science 342, 866-871 (2013).
46. Fransson, S.; Uv, A.; Eriksson, H.; Andersson, M. K.; Wettergren, Y.; Bergo, M.; & Ejeskar, K. p37d is a new isoform of PI3K p110d that increases cell proliferation and is overexpressed in tumors. Oncogene 30, Adv. Online Pub. (2011).
47. Papakonstanti, E. A.; Ridley, A. J.; & Vanhaesebroeck, B. The p110d isoform of PI 3-kinase negatively controls RhoA and PTEN. EMBO J 26, 3050-3061 (2007).
48. Tzenaki, N.; Andreou, M.; Stratigi, K.; Vergetaki, A.; Makrigiannakis, A.; Vanhaesebroeck, B.; & Papakonstanti, E. A. High levels of p110d PI3K expression in solid tumor cells suppress PTEN activity, generating cellular sensitivity to p110d inhibitors through PTEN activation. FASEB J 26, 2498-2508 (2013).
49. Tzenaki, N.; & Papakonstanti, E. A. p110d PI3 kinase pathway: emerging roles in cancer. Front Oncol 3, 40 (2013).
50. Barber, D. F.; Alvarado-Kristensson, M.; Gonzalez-Garcia, A.; Pulido, R.; & Carrera, A. C. PTEN regulation, a novel function for the p85 subunit of Phosphoinositide 3-Kinase. Science STKE 2006, pe49-50 (2006).
51. Downward, J.; & Weigelt, B. Genomic determinants of PI3K pathway inhibitor response in cancer. Front Oncol 2, 109 (2012).
52. Rommel, C.; Camps, M.; & Ji, H. PI3Kd and PI3Kg: Partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol 7, 191-201 (2007).
53. Gamper, C. J.; & Powell, J. D. All PI3Kinase signaling is not mTOR: Dissecting mTOR-dependent and independent signaling pathways in T cells. Front Immunol 3 (2012).
54. Thomson, A. W.; Turnquist, H. R.; & Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 9, 324-337 (2009).
55. Ghigo, A.; Damilano, F.; Braccini, L.; & Hirsch, E. PI3K inhibition in inflammation: Toward tailored therapies for specific diseases. BioEssays 32, 185-196 (2010).
56. Okkenhaug, K.; Ali, K.; & Vanhaesebroeck, B. Antigen receptor signalling: A distinctive role for the p110d isoform of PI3K. Trends Immunol 28, 80-87 (2007).
57. Fruman, D. A.; Snapper, S. B.; Yballe, C. M.; Davidson, L.; Yu, J. Y.; Alt, F. W.; & Cantley, L. C. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85a Science 283, 393-397 (1999).
58. Suzuki, H.; Terauchi, Y.; Fujiwara, M.; Aizawa, S.; Yazaki, Y.; Kadowaki, T.; & Koyasu, S. Xid-like immunodeficiency in mice with disruption of the p85a subunit of phosphoinositide 3-kinase. Science 283, 390-392 (1999).
59. Chen, D.; Mauvais-Jarvis, F.; Bluher, M.; Fisher, S. J.; Jozsi, A.; Goodyear, L. J.; Ueki, K.; & al. p50a/p55a phosphoinositide 3-kinase knockout mice exhibit enhanced insulin sensitivity. Mol Cell Biol 24, 320-329 (2004).
60. Deane, J. A.; Trifilo, M. J.; Yballe, C. M.; Choi, S.; Lane, T. E.; & Fruman, D. A. Enhanced T cell proliferation in mice lacking the p85b subunit of phosphoinositide 3-kinase. J Immunol 172, 6615-6625 (2004).
61. Matheu, M. P.; Deane, J. A.; Parker, I.; Fruman, D. A.; & Cahalan, M. D. Class IA phosphoinositide 3-kinase modulates basal lymphocyte motility in the lymph node. J Immunol 179, 2261-2269 (2007).
62. Suire, S.; Condliffe, A. M.; Ferguson, G. J.; Ellson, C. D.; Guillou, H.; Davidson, K.; Welch, H.; & al. Gbgs and the Ras binding domain of p110g are both important regulators of PI3Kg signalling in neutrophils. Nat Cell Biol 8, 1303-1309 (2006).
63. Ramadani, F.; Bolland, D. J.; Garcon, F.; Emery, J. L.; Vanhaesebroeck, B.; Corcoran, A. E.; & Okkenhaug, K. The PI3K isoforms p110a and p110d are essential for pre-B cell receptor signaling and B cell development. Sci Signal 3, ra60 (2010).
64. Clayton, E.; Bardi, G.; Bell, S. E.; Chantry, D.; Downes, C. P.; Gray, A.; Humphries, L. A.; & al. A crucial role for the p110d subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med 196, 753-763 (2002).
65. Jou, S.-T.; Carpino, N.; Takahashi, Y.; Piekorz, R.; Chao, J.-R.; Carpino, N.; Wang, D.; & al. Essential, nonredundant role for the phosphoinositide 3-kinase p110d in signaling by the B-cell receptor complex. Mol Cell Biol 22, 8580-8591 (2002).
66. Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; & al. Impaired B and T cell antigen receptor signaling in p110d PI 3-kinase mutant mice. Science 297, 1031-1034 (2002).
67. Zhang, T.-t.; Okkenhaug, K.; Nashed, B. F.; Puri, K. D.; Knight, Z. A.; Shokat, K. M.; Vanhaesebroeck, B.; & al. Genetic or pharmaceutical blockade of p110d phosphoinositide 3-kinase enhances IgE production. J Allergy Clin Immunol 122, 811-819.e812 (2008).
68. Zhang, T.-t.; Makondo, K. J.; & Marshall, A. J. p110d phosphoinositide 3-kinase represses IgE switch by potentiating BCL6 expression. J Immunol 188, 3700-3708 (2012).
69. Sasaki, T.; Irie-Sasaki, J.; Jones, R. G.; Oliveira-dos-Santos, A. J.; Stanford, W. L.; Bolon, B.; Wakeham, A.; & al. Function of PI3Kg in thymocyte development, T cell activation, and neutrophil migration. Science 287, 1040-1046 (2000).
70. Ji, H.; Rintelen, F.; Waltzinger, C.; Bertschy Meier, D.; Bilancio, A.; Pearce, W.; Hirsch, E.; & al. Inactivation of PI3Kg and PI3Kd distorts T-cell development and causes multiple organ inflammation. Blood 110, 2940-2947 (2007).
71. Alcazar, I.; Cortes, I.; Zaballos, A.; Hernandez, C.; Fruman, D. A.; Barber, D. F.; & Carrera, A. C. p85b phosphoinositide 3-kinase regulates CD28 coreceptor function. Blood 113, 3198-3208 (2009).
72. Oak, J. S.; Deane, J. A.; Kharas, M. G.; Luo, J.; Lane, T. E.; Cantley, L. C.; & Fruman, D. A. Sjögren's syndrome-like disease in mice with T cells lacking class 1A phosphoinositide-3-kinase. Proc Natl Acad Sci USA 103, 16882-16887 (2006).
73. Deane, J. A.; Kharas, M. G.; Oak, J. S.; Stiles, L. N.; Luo, J.; Moore, T. I.; Ji, H.; & al. T-cell function is partially maintained in the absence of class IA phosphoinositide 3-kinase signaling. Blood 109, 2894-2902 (2007).
74. Rolf, J.; Bell, S. E.; Kovesdi, D.; Janas, M. L.; Soond, D. R.; Webb, L. M. C.; Santinelli, S.; & al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol 185, 4042-4052 (2010).
75. Rodríguez-Borlado, L.; Barber, D. F.; Hernández, C.; Rodríguez-Marcos, M. A.; Sánchez, A.; Hirsch, E.; Wymann, M.; & al. Phosphatidylinositol 3-kinase regulates the CD4/CD8 T cell differentiation ratio. J Immunol 170, 4475-4482 (2003).
76. Garçon, F.; Patton, D. T.; Emery, J. L.; Hirsch, E.; Rottapel, R.; Sasaki, T.; & Okkenhaug, K. CD28 provides T-cell costimulation and enhances PI3K activity at the immune synapse independently of its capacity to interact with the p85/p110 heterodimer. Blood 111, 1464-1471 (2008).
77. Thomas, M. S.; Mitchell, J. S.; DeNucci, C. C.; Martin, A. L.; & Shimizu, Y. The p110g isoform of phosphatidylinositol 3-kinase regulates migration of effector CD4 T lymphocytes into peripheral inflammatory sites. J Leukoc Biol 84, 814-823 (2008).
78. Martin, A. L.; Schwartz, M. D.; Jameson, S. C.; & Shimizu, Y. Selective regulation of CD8 effector T cell migration by the p110g isoform of phosphatidylinositol 3-kinase. J Immunol 180, 2081-2088 (2008).
79. Webb, L. M. C.; Vigorito, E.; Wymann, M. P.; Hirsch, E.; & Turner, M. Cutting edge: T cell development requires the combined activities of the p110g and p110d catalytic isoforms of phosphatidylinositol 3-kinase. J Immunol 175, 2783-2787 (2005).
80. Bilancio, A.; Okkenhaug, K.; Camps, M.; Emery, J. L.; Ruckle, T.; Rommel, C.; & Vanhaesebroeck, B. Key role of the p110d isoform of PI3K in B-cell antigen and IL-4 receptor signaling: comparative analysis of genetic and pharmacologic interference with p110d function in B cells. Blood 107, 642-650 (2006).
81. So, L.; Yea, S. S.; Oak, J. S.; Lu, M.; Manmadhan, A.; Ke, Q. H.; Janes, M. R.; & al. Selective inhibition of phosphoinositide 3-kinase p110a preserves lymphocyte function. J Biol Chem 288, 5718-5731 (2013).
82. Aragoneses-Fenoll, L.; Montes-Casado, M.; Herranz, J.; Acosta, Y. Y.; Martínez, S.; Blanco-Aparicio, C.; Pastor, J.; & al. ETP-46321, a dual p110a/d class IA phosphoinositide 3-kinase inhibitor modulates CD28 and ICOS costimulation of CD4+ T lymphocyte responses, cell survival, and immune responses in vivo (Submitted) (2013).
83. Shi, J.; Cinek, T.; Truitt, K. E.; & Imboden, J. B. Wortmannin, a phosphatidylinositol 3-kinase inhibitor, blocks antigen-mediated, but not CD3 monoclonal antibody-induced, activation of murine CD4+ T cells. J Immunol 158, 4688-4695 (1997).
84. Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C. J.; McLauchlan, H.; Klevernic, I.; & al. The selectivity of protein kinase inhibitors: a further update. Biochem J 408, 297-315 (2007).
85. Okkenhaug, K.; Patton, D. T.; Bilancio, A.; Garcon, F.; Rowan, W. C.; & Vanhaesebroeck, B. The p110d isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol 177, 5122-5128 (2006).
86. Haylock-Jacobs, S.; Comerford, I.; Bunting, M.; Kara, E.; Townley, S.; Klingler-Hoffmann, M.; Vanhaesebroeck, B.; & al. PI3Kd drives the pathogenesis of experimental autoimmune encephalomyelitis by inhibiting effector T cell apoptosis and promoting Th17 differentiation. J Autoimmun 36, 278-287 (2011).
87. Soond, D. R.; Bjorgo, E.; Moltu, K.; Dale, V. Q.; Patton, D. T.; Torgersen, K. M.; Galleway, F.; & al. PI3K p110d regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood 115, 2203-2213 (2010).
88. Rolf, J.; Fairfax, K.; & Turner, M. Signaling pathways in T follicular helper cells. J Immunol 184, 6563-6568 (2010).
89. Gigoux, M.; Shang, J.; Pak, Y.; Xu, M.; Choe, J.; Mak, T. W.; & Suh, W.-K. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc Natl Acad Sci USA 106, 20371-20376 (2009).
90. Xu, H.; Li, X.; Liu, D.; Li, J.; Zhang, X.; Chen, X.; Hou, S.; & al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 496, 523-527 (2013).
91. Nukada, Y.; Okamoto, N.; Konakahara, S.; Tezuka, K.; Ohashi, K.; Mizuno, K.; & Tsuji, T. AILIM/ICOS-mediated elongation of activated T cells is regulated by both the PI3-kinase/Akt and Rho family cascade. Int Immunol 18, 1815-1824 (2006).
92. Salazar-Fontana, L. I.; Barr, V.; Samelson, L. E.; & Bierer, B. E. CD28 engagement promotes actin polymerization through the activation of the small Rho GTPase Cdc42 in human T cells. J Immunol 171, 2225-2232 (2003).
93. Franko, J. L.; & Levine, A. D. Antigen-independent adhesion and cell spreading by inducible costimulator engagement inhibits T cell migration in a PI-3K-dependent manner. J Leukoc Biol 85, 526-538 (2009).
94. Acosta, Y. Y.; Ojeda, G.; Zafra, M. P.; Seren Bernardone, I.; Sánchez, A.; Dianzani, U.; Portolés, P.; & al. Dissociation of actin polymerization and lipid raft accumulation by ligation of the Inducible Costimulator (ICOS, CD278). Inmunología 31, 4-12 (2012).
95. Schneider, H.; Valk, E.; da Rocha Dias, S.; Wei, B.; & Rudd, C. E. CTLA-4 up-regulation of lymphocyte function-associated antigen 1 adhesion and clustering as an alternate basis for coreceptor function. Proc Natl Acad Sci USA 102, 12861-12866 (2005).
96. Patton, D. T.; Garden, O. A.; Pearce, W. P.; Clough, L. E.; Monk, C. R.; Leung, E.; Rowan, W. C.; & al. Cutting Edge: The phosphoinositide 3-kinase p110d is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol 177, 6598-6602 (2006).
97. Liu, D.; Zhang, T.; Marshall, A. J.; Okkenhaug, K.; Vanhaesebroeck, B.; & Uzonna, J. E. The p110d isoform of Phosphatidylinositol 3-Kinase controls susceptibility to Leishmania major by regulating expansion and tissue homing of regulatory T cells. J Immunol 183, 1921-1933 (2009).
98. Sauer, S.; Bruno, L.; Hertweck, A.; Finlay, D.; Leleu, M.; Spivakov, M.; Knight, Z. A.; & al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA 105, 7797-7802 (2008).
99. Merkenschlager, M.; & von Boehmer, H. PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. J Exp Med 207, 1347-1350 (2010).
100. Walsh, P. T.; Buckler, J. L.; Zhang, J.; Gelman, A. E.; Dalton, N. M.; Taylor, D. K.; Bensinger, S. J.; & al. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J Clin Invest 116, 2521-2531 (2006).
101. Ni, H.-T.; Deeths, M. J.; & Mescher, M. F. Phosphatidylinositol 3 kinase activity is not essential for B7-1-mediated costimulation of proliferation or development of cytotoxicity in murine T cells. J Immunol 157, 2243-2246 (1996).
102. Phu, T.; Haeryfar, S. M. M.; Musgrave, B. L.; & Hoskin, D. W. Phosphatidylinositol 3-kinase inhibitors prevent mouse cytotoxic T-cell development in vitro. J Leukoc Biol 69, 803-814 (2001).
103. Macintyre, A. N.; Finlay, D.; Preston, G.; Sinclair, L. V.; Waugh, C. M.; Tamas, P.; Feijoo, C.; & al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 34, 224-236 (2011).
104. Camps, M.; Ruckle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; & al. Blockade of PI3Kg suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Medicine 11, 936-943 (2005).
105. Randis, T. M.; Puri, K. D.; Zhou, H.; & Diacovo, T. G. Role of PI3Kδ and PI3Kγ in inflammatory arthritis and tissue localization of neutrophils. Eur J Immunol 38, 1215-1224 (2008).
106. Haruta, K.; Mori, S.; Tamura, N.; Sasaki, A.; Nagamine, M.; Yaguchi, S.-i.; Kamachi, F.; & al. Inhibitory effects of ZSTK474, a phosphatidylinositol 3-kinase inhibitor, on adjuvant-induced arthritis in rats. Inflamm Res 61, 551-562 (2012).
107. Hayer, S.; Pundt, N.; Peters, M. A.; Wunrau, C.; Kühnel, I.; Neugebauer, K.; Strietholt, S.; & al. PI3Kg regulates cartilage damage in chronic inflammatory arthritis. FASEB J 23, 4288-4298 (2009).
108. Bartok, B.; Boyle, D. L.; Liu, Y.; Ren, P.; Ball, S. T.; Bugbee, W. D.; Rommel, C.; & al. PI3 kinase d is a key regulator of synoviocyte function in rheumatoid arthritis. Amer J Pathol 180, 1906-1916 (2012).
109. Vinuesa, C. G.; Cook, M. C.; Angelucci, C.; Athanasopoulos, V.; Rui, L.; Hill, K. M.; Yu, D.; & al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435, 452-458 (2005).
110. Yu, D.; Tan, A. H.-M.; Hu, X.; Athanasopoulos, V.; Simpson, N.; Silva, D. G.; Hutloff, A.; & al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature 450, 299-303 (2007).
111. Lee, S. K.; Silva, Diego G.; Martin, Jaime L.; Pratama, A.; Hu, X.; Chang, P.-P.; Walters, G.; & al. Interferon-g excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity 37, 880-892 (2012).
112. Sweet, R. A.; Lee, S. K.; & Vinuesa, C. G. Developing connections amongst key cytokines and dysregulated germinal centers in autoimmunity. Curr Opin Immunol 24, 658-664 (2012).
113. Barber, D. F.; Bartolome, A.; Hernandez, C.; Flores, J. M.; Redondo, C.; Fernandez-Arias, C.; Camps, M.; & al. PI3Kg inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Medicine 11, 933-935 (2005).
114. Barber, D. F.; Bartolome, A.; Hernandez, C.; Flores, J. M.; Fernandez-Arias, C.; Rodriguez-Borlado, L.; Hirsch, E.; & al. Class IB-phosphatidylinositol 3-kinase (PI3K) deficiency ameliorates IA-PI3K-induced systemic lupus but not T cell invasion. J Immunol 176, 589-593 (2006).
115. Suárez-Fueyo, A.; Barber, D. F.; Martínez-Ara, J.; Zea-Mendoza, A. C.; & Carrera, A. C. Enhanced phosphoinositide 3-kinase d activity is a frequent event in Systemic Lupus Erythematosus that confers resistance to activation-induced T cell death. J Immunol 187, 2376-2385 (2011).
116. Maxwell, M. J.; Tsantikos, E.; Kong, A. M.; Vanhaesebroeck, B.; Tarlinton, D. M.; & Hibbs, M. L. Attenuation of phosphoinositide 3-kinase d signaling restrains autoimmune disease. J Autoimmun 38, 381-391 (2012).
117. Acosta, Y. Y.; Montes-Casado, M.; Aragoneses-Fenoll, L.; Dianzani, U.; Portolés, P.; & Rojo, J. M. Suppression of CD4+ T lymphocyte activation “in vitro” and experimental encephalomyelitis “in vivo” by the phosphatidyl inositol 3-kinase inhibitor PIK-75. Int J Immunopathol Pharmacol MS 1814, (Enviado) (2013).
118. Dagia, N. M.; Agarwal, G.; Kamath, D. V.; Chetrapal-Kunwar, A.; Gupte, R. D.; Jadhav, M. G.; Dadarkar, S. S.; & al. A preferential p110a/g PI3K inhibitor attenuates experimental inflammation by suppressing the production of proinflammatory mediators in a NF-kB-dependent manner. Am J Physiol Cell Physiol 298, C929-941 (2010).
119. Roller, A.; Perino, A.; Dapavo, P.; Soro, E.; Okkenhaug, K.; Hirsch, E.; & Ji, H. Blockade of phosphatidylinositol 3-kinase (PI3K)δ or PI3Kγ reduces IL-17 and ameliorates Imiquimod-induced psoriasis-like dermatitis J Immunol 189, 4612-4620 (2012).
120. Schreiber, R. D.; Old, L. J.; & Smyth, M. J. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science 331, 1565-1570 (2011).
121. Vesely, M. D.; Kershaw, M. H.; Schreiber, R. D.; & Smyth, M. J. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29, 235-271 (2011).
122. Zou, W.; & Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol 8, 467-477 (2008).
123. Strauss, L.; Bergmann, C.; Szczepanski, M. J.; Lang, S.; Kirkwood, J. M.; & Whiteside, T. L. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: Implications and impact on tumor-mediated immune suppression. J Immunol 180, 2967-2980 (2008).
124. Chen, X. L.; Cao, X. D.; Kang, A. J.; Wang, K. M.; Su, B. S.; & Wang, Y. L. In situ expression and significance of B7 costimulatory molecules within tissues of human gastric carcinoma. World J Gastroenterol 9, 1370-1373 (2003).
125. Martin-Orozco, N.; Li, Y.; Wang, Y.; Liu, S.; Hwu, P.; Liu, Y.-J.; Dong, C.; & al. Melanoma cells express ICOS ligand to promote the activation and expansion of T-regulatory cells. Cancer Res 70, 9581-9590 (2010).
126. Conrad, C.; & Gilliet, M. Plasmacytoid dendritic cells and regulatory T cells in the tumor microenvironment: A dangerous liaison. Oncoimmunology 2, e23887 (2013).
127. Schreiner, B.; Wischhusen, J.; Mitsdoerffer, M.; Schneider, D.; Bornemann, A.; Melms, A.; Tolosa, E.; & al. Expression of the B7-related molecule ICOSL by human glioma cells in vitro and in vivo. Glia 44, 296-301 (2003).
128. Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D. M.; & Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer 7, 95-106 (2007).
129. Fruman, D. A.; & Rommel, C. PI3Kd inhibitors in cancer: Rationale and serendipity merge in the clinic. Cancer Discov 1, 562-572 (2011).
Addenda: Abbreviations and list of PI3K inhibitors
Abbreviations
ABD Adaptor binding domain
AIA Adjuvant-induced arthritis
Akt Protein kinase B
ATG13 Autophagy-related protein 13
ATM Ataxia-telangiectasia mutated protein (ATM)
ATR Ataxia- and Rad3-related protein (ATR)
BAD Bcl-2-associated death promoter
BCAP B-cell adaptor for PI3K
BCL6 B cell lymphoma 6 transcription repressor
BCR B cell receptor, antigen receptor of B lymphocytes
BH BCR-homology GTPase activation domain
Btk Bruton’s tyrosine kinase
BTLA B-and T-lymphocyte attenuator, CD272
CIA Collagen-induced arthritis
CNS Central nervous system
CTLA-4 Cytotoxic T-Lymphocyte Antigen 4, CD152
DNA-PKcs DNA-dependent protein kinase catalytic subunit
EAE Experimental Autoimmune Encephalomyelitis
Erk Extracellular signal-regulated kinase
FLS Fibroblast-like synoviocyte
FYVE Fab 1 (yeast orthologue of PIKfyve), YOTB, Vac 1 (vesicle transport protein), and EEA1 domain
FOXO forkhead transcription factor/forkhead box O transcription factors
GAP GTPase activating factors
GC Germinal center
GEF Guanine nucleotide exchange factors
G proteins Heterotrimeric Guanine nucleotide-binding proteins
GPCR G protein–coupled receptor
GSK-3 Glycogen synthase kinase 3
ICOS Inducible costimulator, CD278
ITAM Immunoreceptor Tyrosine-based activation motif
Itk Interleukin-2-inducible T-cell tyrosine kinase
IKK IkB kinase
HEAT Huntington, Elongation Factor 3, PR65/A, TOR domains
LAT Linker of activation of T cells
MAPK Mitogen activated protein kinase
MHC Major Histocompatibility Complex
MOG Myelin Oligodendrocyte Glycoprotein
MS Multiple sclerosis
mTOR Mechanistic Target of Rapamycin
mTORC1 Complex of mTOR containing Raptor
mTORC2 Complex of mTOR containing Rictor
NFAT Nuclear factor of activated T-cells
NF-kB Nuclear factor k-light-chain-enhancer of activated B cells
NK Natural killer lymphocytes
S6K p70 S6 kinase
PD-1 Programmed death-1, CD279
PDGF Platelet-derived growth factor
PDK1 Phosphoinositide-dependent kinase 1
PGE2 Prostaglandin E2
PH Pleckstrin homology domain
PIKK PI3K-like protein kinases
PI3K Phosphoinositide-3 kinases (PI3Ks)
PKCq Protein kinase q
PKB Protein kinase B
PLCg Phospholipase C g
PROPPIN b-propeller that bind phosphoinositide species
PTEN Phosphatase and Tensin Homologue
PtdInsP Phosphorylated inositol lipids
PX Phox homology domain
RHEB Ras homologue enriched in brain
SH2 Src homology region 2 domain
SH3 Src homology region 3 domain
SHP-1 SH2 domain-containing Inositol Phosphatase-1
SHP-2 SH2 domain-containing Inositol Phosphatase-2
SGK1 Serum- and glucocorticoid-inducible kinase (SGK) 1
SLE Systemic Lupus Erythematosus
TCR T cell receptor, antigen receptor of T lymphocytes
Tfh Follicular helper T cells
TNF Tumor necrosis factor
TSC1, TSC2 Tuberous sclerosis complex proteins 1 and 2
Treg CD4+ regulatory T lymphocytes
TRIM T cell receptor (TCR)-interacting molecule
WD repeat WD40 repeat, b-transducin repeat
PI3K inhibitors
A66 PI3K inhibitor, p110a
AS-605240 PI3K inhibitor, p110g
AS-604850 PI3K inhibitor, p110a/g
CAL-101 PI3K inhibitor, p110d
ETP-46321 PI3K inhibitor, p110a/d
GDC-0941 PI3K inhibitor, pan-PI3K
IC87114 PI3K inhibitor, p110d
INK007 PI3K inhibitor, p110d
LY294002 PI3K inhibitor, broad-spectrum
MLN1117 PI3K inhibitor, p110a
PI-103 PI3K inhibitor, broad-spectrum
PIK-75 PI3K inhibitor, p110a/DNA-PK inhibitor
PIK-90 PI3K inhibitor, broad-spectrum
TGX-221 PI3K inhibitor, p110b
Wortmannin PI3K inhibitor, broad-spectrum
ZSTK474 PI3K inhibitor, broad-spectrum