REVISIÓN |
SREBP-1c, ChREBP y LXR: Su influencia en el desarrollo del hígado graso no alcohólico
Elvira López-Oliva Muñoz1, Emilia Muñoz Martínez2*
1Sección departamental de Fisiología, Facultad de Farmacia, Universidad Complutense, Pza. Ramón y Cajal s/n, 28040, Madrid. 2Académica Correspondiente de la Real Academia Nacional de Farmacia.
*e-mail: emilia@ucm.es
Recibido el 11 de diciembre de 2013 -An. Real Acad. Farm. Vol. 80, Nº 1 (2014), pág. 14-48
RESUMEN
El hígado graso no alcohólico (HGNA) es una enfermedad que se define como un espectro continuo que oscila entre una esteatosis macrovesicular, de curso clínico favorable, hasta un cuadro de esteatohepatitis no alcohólica (EHNA), que da lugar a daños irreversibles y que se ha convertido en los últimos años en un problema sanitario de primera magnitud. El HGNA se caracteriza por una infiltración grasa de los hepatocitos que se asocia con un estado de resistencia a la insulina y por ello ligado, como factor de riesgo, al síndrome metabólico. Los factores de transcripción SREBP-1c (proteína de unión al elemento regulador del esterol), ChREBP (proteína de unión al elemento de respuesta a carbohidratos) y LXR (receptor X hepático) son reguladores fundamentales de la homeostasis lipídica y glucídica y de la inflamación, cuya activación regula al alza genes implicados en la síntesis de novo de los ácidos grasos en respuesta a insulina, glucosa y oxiesteroles, tanto en condiciones fisiológicas como patológicas. En esta revisión se describen datos recientes sobre la biología, la regulación y la coordinación funcional entre SREBP-1c, ChREBP y LXR y su relación con el HGNA. El desarrollo de agonistas selectivos de estos factores les hacen ser prometedoras dianas en el tratamiento del HGNA y de la EHNA. |
Palabras clave: Hígado graso no alcohólico; Factores de trascripción; Lipogénesis.
aBSTRACT
SREBP-1c, ChREBP and LXR: Their role in the pathogenesis of the non-alcoholic fatty liver.
Nonalcoholic fatty liver disease (NAFLD) has become a major public health issue that comprises a disease spectrum which ranges from benign hepatic steatosis to non-alcoholic steatohepatitis (NASH), leading to irreversible liver damages. Deposition of excess triglycerides within liver cells is the hallmark of NAFLD, which is associated with a loss of insulin sensitivity. A growing body of evidence implicates the lipogenic transcription factors SREBP-1c (sterol regulatory element-binding protein), ChREBP (carbohydrate responsive element-binding protein) and LXR (liver X receptor) in the pathogenesis of NAFLD. These factors have emerged as central regulators of the de novo fatty acid synthesis, the glucose homeostasis and the inflammation in response to insulin, glucose and oxysterols, under both physiological and physiopathological conditions. In this review we describe recent findings in the biology, the function and the cross-regulation between SREBP-1c, ChREBP and LXR on the control of lipid and glucose metabolism and their link to NAFLD. Specific pharmacologic ligands are available, making them attractive therapeutic targets for NAFLD and NASH. |
Keywords: Non-alcoholic fatty liver; Transcription factors; Lipogenesis.
INTRODUCCIÓN
Los triglicéridos (TG) almacenados en los adipocitos constituyen una forma de depósito de nutrientes fácilmente disponible, ante fluctuaciones de su disponibilidad y de la demanda energética. Solo cuando la capacidad de almacenar grasa del tejido adiposo blanco (TAB) se ve sobrepasada (dietas altas en grasa (HFD) o en carbohidratos (HCD), estados de resistencia a la insulina (RI), etc.), se produce un depósito anómalo en tejidos no adiposos, como el hígado, alterando el balance entre el aporte de lípidos (ácidos grasos libres (AGL) circulantes captados y los sintetizados de novo) y su catabolismo (oxidación de ácidos grasos (AG) y/o secreción de lipoproteínas de muy baja densidad (VLDL) (1). Ello da lugar a una alteración del metabolismo lipídico hepático, que puede tener consecuencias clínicas graves.
El hígado graso no alcohólico se caracteriza histológicamente por una infiltración grasa en el parénquima hepático (>55mg/g), en individuos que no consumen alcohol (<20 ó 40g/semana para hombres o mujeres, respectivamente) o que no padecen enfermedades virales, congénitas o autoinmunes del hígado (2). Desde que fue descrito por Ludwig y col en 1980 (3), el hígado graso no alcohólico (HGNA) se define como un espectro continuo que va incrementando su gravedad, desde una esteatosis simple macrovesicular (HGNA), de curso clínico favorable, hasta una esteatohepatitis no alcohólica (EHNA) caracterizada por la existencia de necroinflamación, apoptosis de los hepatocitos, fibrosis, cirrosis y eventualmente carcinoma hepatocelular, lesiones indistinguibles de las del hígado graso alcohólico (4,5). Debido a que el acúmulo de TG hepáticos está asociado a un estado de resistencia a la insulina, el HGNA es considerado como la manifestación hepática del síndrome metabólico y ligado muy estrechamente, como factor de riesgo, a todas sus manifestaciones: obesidad, diabetes tipo 2, dislipemia, hipertensión y aterosclerosis (6,7). Por ello, aunque la incidencia del HGNA es alta entre la población (20-30 % en los adultos y 3-10% en los niños), según cifras de EEUU extrapolables a Europa y a España (8), la obesidad (80-90%) y la diabetes tipo 2 (30-50%) acentúan el problema, pudiendo desarrollar EHNA hasta en el 35% de los casos (9). Así pues, el predominio de los hábitos sedentarios, la ingesta hipercalóríca y la prevalencia de obesidad en la población occidental han aumentado el riesgo de padecer EHNA, por lo que se ha convertido en la causa más común de enfermedad crónica hepática, configurándose como un importante problema económico y de salud pública (10), con un índice de mortalidad muy elevado (11). En la clínica, además de las pruebas de laboratorio, existen técnicas no invasivas de evaluación diagnóstica (ultrasonidos, resonancia magnética, tomografía computarizada etc.), pero sin embargo, el cuadro histopatológico real y su gravedad, solo puede establecerse mediante la biopsia hepática (5). En cuanto al tratamiento, aunque se han postulado el uso de agentes sensibilizadores a la insulina (metformina, tiazolidinedionas), antilipémicos (fibratos), antioxidantes, inhibidores del factor de necrosis tumoral α (TNFα), ácidos grasos poliinsaturados (PUFA) etc., no existe una terapia específica de EHNA. Por ello, el tratamiento recomendado hoy día, se basa en el cambio de estilo de vida y de hábitos dietéticos y en el control de peso (4).
En este trabajo se revisan, en primer término, los mecanismos patogénicos de HGNA y su progresión hacia EHNA. En segundo lugar, se examinan los datos actuales sobre la biología y la función de los factores de transcripción; SREBP-1c (proteína de unión al elemento regulador del esterol), ChREBP (proteína de unión al elemento de respuesta a carbohidratos) y el receptor X hepático (LXR), como reguladores esenciales de la homeostasis lipídica, glucídica y de la inflamación, haciendo énfasis en su implicación en la patogenia del HGNA.
PATOGENIA DEL HIGADO GRASO NO ALCOHÓLICO
La patogenia del hígado graso no alcohólico es multifactorial y todavía no bien conocida. Se han utilizado varios modelos animales que desarrollan síntomas similares, a fin de establecer los mecanismos patogénicos del HGNA y su progresión a la EHNA. Entre ellos, los más usados son los modelos genéticos (ob/ob, con una mutación del gen de leptina y db/db, con una mutación del gen del receptor de leptina) y modelos con EHNA inducida por dieta (HFD, HCD, alta en fructosa, deficiente en colina-metionina, etc. (12).
Resistencia a la insulina en la esteatosis
El origen de la infiltración grasa del hígado es relativamente comprendido. Se acepta hoy que la RI es un factor primordial en el desarrollo y progresión de la enfermedad. Se ha sugerido a este respecto que la RI periférica sería el primer acontecimiento que originaría un aumento del flujo de AGL al hígado, mediante la reducción del efecto supresor de la lipólisis por insulina en el tejido adiposo (Figura 1). En el HGNA, como en otros modelos de RI, el hígado se hace resistente a la acción de la hormona, incrementando la gluconeogénesis y la glucogenólisis (13) mientras que, por el contrario, responde a la hiperinsulinemia aumentando la transcripción de los genes lipogénícos. El aumento en la producción de glucosa hepática acoplada a su menor captación periférica acentúa la hiperglucemia e incrementa la secreción de insulina, aumentando todavía más la lipogénesis hepática (14). Sin embargo, no está claro si la RI hepática es la causa o la consecuencia de la esteatosis (15). Algunos estudios señalan una asociación entre el HGNA y la RI, ya que el decrecimiento de los TG hepáticos se correlaciona con la mejora de la sensibilidad a la insulina (16). Sin embargo, se ha sugerido recientemente que especies lipídicas como los ácidos grasos saturados (AGS), los diacilgliceroles (DAG) o las ceramidas, son los verdaderos determinantes de su desarrollo (17,18) y no los TG, que podrían ejercer una acción protectora, al tamponar el exceso de AG. En este sentido, aunque la relación entre los DAG y la resistencia insulínica no se ha podido confirmar en modelos que sobreexpresan enzimas de la esterificación, como la diacilglicerol-acil-transferasa-2 (DGAT2) (19) o se bloquea la secreción de VLDL (20), en los que se muestra una clara disociación entre esteatosis y RI, en varios modelos de HGNA, no obstante, se ha demostrado que los DAG como intermediarios del metabolismo de los AG inducen RI interfiriendo con la señalización la de insulina, mediante la activación de la proteína-quinasa Cε (PKCε) que fosforila los residuos de serina en los sustratos del receptor de insulina (IRS-1/2), alterando más adelante en la vía, la actividad de las quinasas fosfatidil inositol-3-quinasa (PI3-K) y Akt (21)(22).

Figura 1.- Mecanismos patogénicos de la esteatosis hepática (HGNA) y de su progresión a la esteatohepatitis no alcohólica (EHNA). El hígado graso no alcohólico es una enfermedad que se define como un espectro continuo que incrementa su gravedad desde una esteatosis hepática benigna (HGNA), caracterizada por la infiltración grasa del parénquima hepático, hasta una esteatohepatitis no alcohólica (EHNA), que está determinada por la existencia de necroinflamación, apoptosis de los hepatocitos, fibrosis, cirrosis y en su caso carcinoma hepatocelular. La resistencia a la insulina es un factor primordial en el desarrollo del HGNA, mientras que su progresión a la EHNA se debe a la interacción de múltiples factores, genéticos y ambientales, que ejercen efectos deletéreos en el hepatocito dando lugar al daño tisular. (Véase el texto)
Vías metabólicas implicadas en la esteatosis hepática
La esteatosis hepática se desarrolla como resultado del desequilibrio entre el aporte y la degradación de los lípidos. Los lípidos almacenados en el TAB que fluyen al hígado como AGL, los sintetizados en el hígado mediante la lipogénesis de novo y los AG procedentes de la dieta, son las fuentes lipídicas que, en condiciones fisiológicas, contribuyen a formar la grasa hepática. Después de la esterificación, los TG pueden ser almacenados como gotas lipídicas en los hepatocitos, secretados a la sangre como partículas VLDL, o hidrolizados de nuevo a AG para su oxidación. Por consiguiente, la infiltración grasa del hígado puede producirse por alguna de las siguientes causas: el aumento de la cantidad de AGL que alcanzan el hígado por la porta; el incremento de la lipogénesis de novo y la disminución de la β-oxidación de los AG y/o de la secreción de TG en forma de VLDL. En pacientes con EHNA Donnelly y col (23) demuestran que el 60% de los TG hepáticos proceden de los AGL circulantes, el 25% deriva de la lipogénesis de novo y el 15% restante se forma a partir de los AG procedentes de la dieta (Figura 1). Por lo tanto, el elevado aporte de AGL al hígado y el incremento de la síntesis de novo de los AG, son las principales causas del acúmulo graso en la esteatosis, mientras que la alteración de la vías de oxidación de los AG y/o de la secreción de VLDL, presentan una menor incidencia.
Aporte de AGL al hígado
Como ya se ha indicado, la RI periférica contribuye a aumentar el flujo de AGL liberados desde el TAB hacia el hígado, al reducir la acción supresora de la insulina sobre la lipasa sensible a hormona (24). Un aumento de la capacidad hepática para captar estos AGL circulantes es crucial para inducir el hígado graso. Teniendo en cuenta que la tasa de captación de los AGL, depende de su concentración y de la capacidad de transporte del hepatocito a través de la membrana, la alteración de sus sistemas de transporte influye significativamente en el desarrollo del HGNA. En este sentido, estudios recientes han demostrado que el aumento en la expresión de las proteínas de transporte de AG ((FATPs), la traslocasa de AG (FAT/CD36) y las proteínas ligadoras de AG (FABPs), parecen favorecer la lipotoxicidad y el desarrollo de esteatosis hepática, como se ha observado en el hígado de pacientes con HGNA (25). Por el contrario, la deleción del gen FATP5,disminuye la obesidad, la esteatosis y la RI, en ratones sometidos a una dieta HFD (26).
Lipogénesis de novo
A consecuencia de la ingesta alimenticia, los animales superiores utilizan los carbohidratos preferentemente para la formación de ATP. Cuando la ingesta de carbohidratos es excesiva, la vía de la lipogénesis de novo permite su conversión en AG, que a su vez, pueden ser esterificados a TG en el hígado. En el HGNA el aumento de la glucemia y la hiperinsulinemia derivadas de la RI favorecen, de forma sinérgica, la conversión de glucosa en AG aumentando la expresión de las enzimas glucolíticas y lipogénicas mediante la activación de los factores de transcripción (FT), SREBP-1c, ChREBP y LXR (Figura 2) (27,28). La conversión de glucosa en AG, incluye la entrada del piruvato procedente de la glucólisis al ciclo de Krebs, en la mitocondria. El citrato formado es transportado al citosol donde se convierte a acetil-CoA por la enzima ATP citrato-liasa (ACL). A su vez, la acetil-CoA-carboxilasa (ACC1), la primera enzima limitante de la lipogénesis de novo, convierte el acetil-CoA en malonil-CoA (29), que se transforma en palmitoil-CoA, por acción de la ácido graso-sintasa (FAS) (30). El ácido palmítico puede ser elongado o desaturado mediante la acción de las enzimas, elongasa de larga cadena 6 (ELOLV6) y estearil-CoA-desaturasa-1 (SCD1) (31) y finalmente, las enzimas glicerol-3-fosfato aciltransferasa mitocondrial (GPAT), 1-acilglicerol-3-fosfato aciltransferasas (AGPATs), la lipina 1 y la DGAT2, que catalizan la esterificación de los acil-CoA sobre los carbonos 1, 2 y 3 del glicerol-3-fosfato respectivamente, sintetizan glicerolípidos (32). El receptor nuclear LXR es el regulador fundamental (33), capaz de aumentar la expresión de los FT citoplásmicos SREBP-1c, inducido por insulina (34) y ChREBP, activado por glucosa (35) ambos, blancos directos de LXR. A su vez LXR, SREBP-1c y ChREBP inducen la expresión de los genes lipogénicos y glucolíticos que intervienen en la lipogénesis de novo (33, 34, 35), cuya sobreexpresión incrementa la infiltración grasa en el hígado.
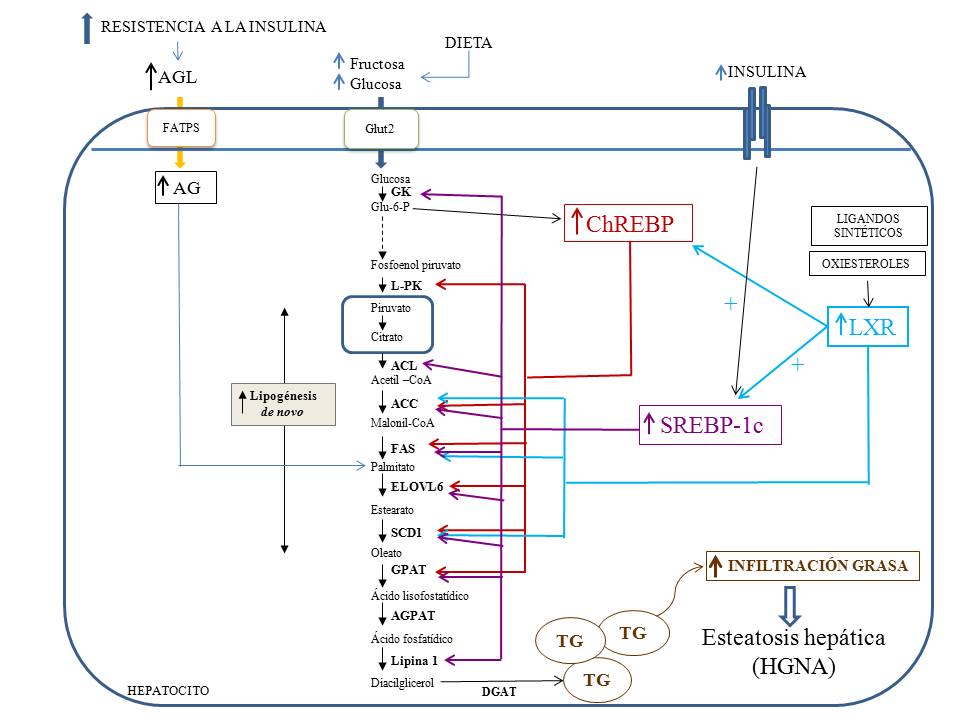
Figura 2.- Interacción funcional entre los factores de transcripción SREBP-1c, ChREBP y LXR en la inducción de la esteatosis hepática. En respuesta a oxiesteroles y otros ligandos, el receptor nuclear LXR, como principal regulador de la lipogénesis de novo, controla los genes que codifican los factores de transcripción SREBP-1c y ChREBP. A su vez, SREBP-1c responde a los altos niveles de insulina, mientras que metabolitos de la glucosa (Glu-6-P, Xu 5P, etc.) que penetra en exceso en el hepatocito mediante el transportador Glut-2, inducen la expresión de ChREBP. Una vez activados, SREBP-1c, ChREBP y LXR incrementan la expresión de genes que codifican enzimas implicadas en la glucólisis (GK, L-PK), en la síntesis de los ácidos grasos (ACL, ACC, FAS, ELOVL6, SCD1) y/o de los triglicéridos (GPAT. Lipina 1), lo que da lugar al aumento del depósito de grasa, que junto a la procedente de los AGL circulantes captados por el hígado, da lugar a la esteatosis. (Véase el texto).
Oxidación de los ácidos grasos
La disminución de las vías de utilización de los AG también puede generar esteatosis hepática. Los AG son catabolizados a través de la ß-oxidación en las mitocondrias y en los peroxisomas y de la ω-oxidación en los microsomas, siendo la ß-oxidación mitocondrial, la vía dominante de metabolización de los AG de cadena corta, media y larga (36). En el HGNA aparecen varios cambios adaptativos dirigidos a incrementar la β-oxidación mitocondrial para compensar la excesiva captación y síntesis de novo de los AG (37). Sin embargo, si se sobrepasa esta capacidad oxidativa, los lípidos pueden acumularse en los hepatocitos. Por ejemplo, un incremento de malonil-CoA puede comprometer la β-oxidación y favorecer el acúmulo de los AG por su función inhibidora de la carnitina-palmitoil transferasa (CPT-1), enzima que regula la entrada de los AG en la mitocondria (38). También una menor actividad oxidativa en los peroxisomas induce una esteatosis severa microvesicular, como se ha observado por deleción del gen del enzima acil-CoA oxidasa (AOX1) en el ratón (39). Por el contrario, la sobreexpresión de los genes de la familia CYP4A de ω-hidroxilasas microsomales, en pacientes con EHNA y en animales alimentados con una dieta HFD, ha sugerido una posible función protectora de estos enzimas sobre la toxicidad lipídica (40), al detoxificar los ácidos dicarboxílicos derivados del exceso de AG, altamente tóxicos.
El receptor nuclear PPARα (receptor activado por proliferadores de peroxisomas α) es el regulador principal de la oxidación de los AG. La activación de PPARα induce la expresión de genes involucrados en la ß y la ω-oxidación (41, 42) y, por ello, su inactivación puede inducir HGNA, como se ha observado en ratones knockout de PPARα en condiciones de ayuno o alimentados con una dieta HFD (43,44). Por el contrario, la estimulación de PPARα con su agonista fenofibrato mejora la esteatosis hepática en el ratón (45).
Secreción de VLDL
El hígado secreta TG en forma de partículas VLDL, que son transportadas hacia los tejidos periféricos y son convertidos en lipoproteínas de baja (LDL) e intermedia (IDL) densidad, por la enzima lipoproteínlipasa. Pacientes con HGNA y EHNA presentan una sobreproducción de VLDL en comparación con sujetos sanos (46), lo que se debe a modificaciones en el ensamblaje y en la secreción de TG. En el HGNA, la disponibilidad de lípidos para el ensamblaje aumenta, a lo que se añade la incapacidad de la insulina para disminuir la producción de VLDL (47). Debido al aumento de TG, la apolipoproteína-B100 (apo-B100) no se degrada y la expresión de la proteína de transferencia microsómica (MPT) se eleva a consecuencia de la sostenida localización de FoxO1 (forkhead box-O1) en el núcleo(48), lo que favorece el aumento de la trigliceridemia observada en estos pacientes. Sin embargo, aunque la exposición crónica a insulina induce mayor producción de VLDL, el incremento de su secreción no compensa la excesiva formación de TG, manteniéndose la esteatosis. Además, la secreción de apo-B100 no se incrementa, lo que sugiere que su producción parece limitar la capacidad del hígado para exportar los TG hepáticos (Figura 1)(48).
PATOGENIA DE EHNA
De esteatosis a esteatohepatitis
El mecanismo de la progresión del HGNA a la EHNA es todavía poco conocido. Se atribuye a la interacción de diferentes factores, tanto genéticos como ambientales, cuya secuencia es desconocida. El modelo tradicional para explicarlo es la hipótesis del doble impacto (“two-hit hypotesis”) (49), que postula una evolución desde la esteatosis, “primer impacto”, en la que el exceso de grasa hace al hígado más susceptible a otras noxas, denominadas en conjunto ”segundo impacto”, que a su vez conducen a la EHNA. En la actualidad, esta teoría está siendo reemplazada por el modelo de lipotoxicidad, que parte de la premisa de que todos los posibles determinantes, metabólicos e inflamatorios de la EHNA no actúan por separado, sino que son interactivos y colaboran en la progresión al daño tisular (50).
Entre los factores que contribuyen a la progresión a la EHNA, la disfunción mitocondrial, el estrés oxidativo y la disminución de la capacidad de defensa antioxidante tienen un papel importante (Figura 1). La aparición de cambios ultraestructurales y funcionales en las mitocondrias de pacientes con EHNA se ha propuesto como un índice de la progresión de la enfermedad (51). También la elevada generación de especies reactivas de oxígeno (ERO), potencialmente tóxicas, por un aumento excesivo de la oxidación de los AG por la vía mitocondrial y /o no mitocondrial (52), puede considerarse como una causa directa de la disfunción de las mitocondrias, puesto que interfieren con la cadena respiratoria y la integridad del DNA mitocondrial (53). Recíprocamente, la disfunción de la cadena de la fosforilación oxidativa y del transporte de electrones puede contribuir al desarrollo de la esteatosis, al inducir la inhibición de la ß-oxidación (54). El aumento de productos de la lipoperoxidación (LPO), como el malondialdehido (MDA) y el 4-hidroxinonenal (4HNE)(55), amplifican el efecto de las ERO al dañar las membranas e inactivar las macromoléculas celulares, lo que intensifica la disfunción mitocondrial y perpetúa la producción de ERO. El aumento consecutivo de la permeabilidad de la membrana externa mitocondrial permite la liberación al citosol de proteínas proapoptóticas, que inducen la muerte de los hepatocitos y favorecen el desarrollo de la inflamación y la fibrosis (56). Las ERO y los productos de LPO, inducen también la síntesis de citoquinas proinflamatorias como TNFα, TGF-ß (factor de crecimiento transformante ß) y las interleucinas IL-6 y IL-8 (57), a través de la activación de las vías del factor nuclear Kappaß/I kappaß quinasa (NF-KB/IKKβ) y de la proteína activadora 1/c-Jun N-terminal quinasa 1 (AP-1/JNK1) propiciando, además del mantenimiento de la RI, la muerte celular (TNFα;TGF-ß), la quimiotaxis de los neutrófilos (IL-8), la formación del infiltrado inflamatorio y la activación de las células de Kupffer (58). El reclutamiento y la activación de las células de Kupffer, que pasan de un fenotipo de macrófagos antiinflamatorios (M2) a un fenotipo proinflamatorio (M1), es crítico para la propagación de la inflamación y del daño tisular (59). Las células de Kupffer también inducen fibrosis mediante la estimulación paracrina de las células estrelladas (HSC), las cuales proliferan y se activan, induciendo su transformación en miofibroblastos profibrogénicos, que incrementan la matriz extracelular al sintetizar colágeno, proteoglicanos e hialuronato (60).
El estrés del retículo endoplásmico (RE) es un mecanismo recientemente implicado en la patogenia y en la progresión de EHNA (Figura 1) (61,62). Cualquier alteración en la homeostasis del RE que afecte la capacidad de plegado de las proteínas, induciendo su acúmulo en el lumen, conduce al estrés del RE. Esta situación pone en marcha una respuesta fisiológica, la respuesta de proteínas no plegadas (UPR), que permite disminuir la carga proteica y aumentar su plegado y su degradación, a través de la activación de una cascada de señales que aumentan la transcripción de chaperonas residentes en el RE (63). Factores como la hipoxia, la exposición a AGS de cadena larga, la hiperinsulinemia, el desequilibrio cálcico o alteraciones en la glicosilación, entre otros, pueden desencadenar la UPR (64). Sin embargo, una UPR insuficiente o inadecuada activa vías de señalización que derivan en el establecimiento de varios determinantes de la progresión a EHNA: la RI (vía inositol requiring enzyme 1α (IRE1α)-JNK1), el estrés oxidativo y la disfunción mitocondrial (vía nuclear factor erythroid-2 related factor 2 (Nrf2), la apoptosis (vía intrínseca) y la inflamación (vía NF-κβ) (63). En este sentido, se ha observado que el exceso de AGS induce estrés del RE por activación de mediadores de la UPR, lo que deriva en daño hepático y apoptosis de los hepatocitos (65).
El desequilibrio en la razón adipoquinas/citoquinas secretadas por el TAB en estados de RI, como la obesidad, tiene un papel esencial en la modulación de la señalización insulínica y en la inflamación (Figura 1). En pacientes con HGNA, además de las citoquinas proinflamatorias, TNFα e IL-6, (66), la alteración de las adipoquinas, también deriva en la progresión a la EHNA. La adiponectina es reconocida por los receptores, AdipoR1 y AdipoR2, expresados en el hígado (67), donde aumenta la β-oxidación de los AG y la sensibilidad a la insulina, mediante la activación de PPARα y la fosforilación de la quinasa activada por AMP (AMPK) (68). Además, la adiponectina modula la inflamación al suprimir la activación de IKKβ inducida por TNFα (69). Por ello, el tratamiento con adiponectina disminuye la esteatosis hepática en el ratón obeso (70), mientras que en pacientes obesos con la EHNA, la reducción de la adiponectinemia se asocia con el grado de esteatosis, de necroinflamación y de fibrosis (71). Por el contrario, los altos niveles de resistina encontrados en plasma de enfermos de EHNA se correlacionan con la RI, la esteatosis y la inflamación (72). La leptina, por su parte, presenta datos contradictorios pues si bien sensibiliza los tejidos a la insulina y activa la ß-oxidación de los AG, parece inducir la inflamación y la fibrogénesis, actuando directamente sobre las células de Kupffer y las células estrelladas del hígado (73).
La RI per se debe ser también considerada como un inductor del daño hepático, debido a su capacidad de aumentar los mediadores inflamatorios y tener efectos directos sobre las células HSC (Figura 1) (74). Además, la lipotoxicidad de las especies lipídicas, que pueden inducir todos los mecanismos desencadenantes del daño tisular ya mencionados, constituyen un factor de primera importancia en la progresión del HGNA a la EHNA (75). Los factores genéticos, por su parte, tienen una gran influencia en su desarrollo. Por ejemplo, el polimorfismo rs738409, que corresponde a una mutación del gen que codifica la proteína fosfolipasa 3 similar a patatina (PNPLA3), también denominada adiponutrina (una proteína multifuncional unida a membrana con actividades lipolíticas y lipogénicas), está significativamente asociada a la presencia de necroinflamación hepática (76). Por último, los endocannabinoides, la proteína de unión al retinol (RBP) y la dehidroepiandrosterona (DHEA), así como mediadores moleculares, tales como el receptor Toll-like 4 (TLR-4), la serotonina y el sistema renina-angiotensina, se han descrito como potenciales mediadores del HGNA y de su progresión a EHNA (77).
SREBP-1c, ChREBP Y LXR.: SU INFLUENCIA EN LA PATOGENIA DEL HGNA.
La homeostasis del metabolismo lipídico requiere de sensores intracelulares que puedan sensibilizarse a cambios hormonales y metabólicos y coordinen las vías metabólicas implicadas. SREBP-1c, CHREBP y LXR, juegan un papel esencial como reguladores de la lipogénesis en respuesta a insulina, glucosa y oxiesteroles y, por ello, su disfunción condiciona la aparición y el desarrollo del HGNA.
SREBPs
Los factores de transcripción SREBPs son miembros de la familia de proteínas cuyo dominio N-terminal (con función de transcripción) presenta una estructura básica/hélice-bucle-hélice/cremallera de leucina (bHLH-LZ) (78). Se han descrito tres miembros de esta familia, SREBP-1a, -1c y -2. SREBP-1a y -1c están codificados por el mismo gen (SREBP-1) a través de transcripción alternativa, diferenciándose en el exon 1 (79), mientras SREBP-2 procede del gen SREBP-2 y tiene un 50% de homología con SREBP-1 (78). Ambas isoformas, SREBP-1a y SREBP-1c, regulan preferentemente la síntesis de los AG y de los TG (80), aunque SREBP-1c es el subtipo predominante en el hígado y en el TAB. En respuesta a insulina, SREBP-1c induce la expresión de los genes que codifican las enzimas lipogénicas ACL, ACC, FAS, ELOVL6, SCD1, GPAT y lipina1) y glucolíticas (glucoquinasa, (GK)) (81), por su capacidad de interacción con los elementos de respuesta al esterol (SRE), ubicados en su promotor (Figura 2). SREBP-2, por su parte, se expresa de forma ubicua y abundante y, preferentemente, controla genes del metabolismo del colesterol (COL) (78,82).
Activación de SREBP-1c
SREBP-1c, como todos los miembros de la familia, se sintetizan como proteínas de membrana del RE, de cuyo emplazamiento deben ser liberados hacia el Golgi donde son activados por proteólisis. Ya en forma madura emigran al núcleo ejerciendo su función de transcripción. El proceso tiene varios pasos: inmediatamente después de su síntesis, la forma precursora de las proteínas SREBPs (pre-SREBPs) ligadas al RE se une a la proteína SCAP (SREBP cleavage-activating protein), situada también en el RE, que actúa como un sensor del COL. En condiciones de COL intracelular bajo, SCAP es requerida para su unión a las proteínas COPII que rodean a las vesículas de transporte, permitiendo al complejo SREBPs-SCAP desplazarse al aparato de Golgi. Una vez en el Golgi, las proteasas Site-1(S1P) y Site-2 (S2P) liberan el extremo N-terminal de las proteínas precursoras, que migran al núcleo como SREBPs maduras (nSREBPs), activando sus genes blanco por unión al SRE en su promotor. Por el contrario, cuando el COL se acumula en la célula, se une a su sensor SCAP por su dominio sensible al esterol, cambiando su conformación molecular y permitiendo su unión a Insig (Insulin-induced gene), otra proteína del RE. Esta unión impide a SCAP unirse a las proteínas COPII, bloqueando la migración del complejo SREBP-SCAP al Golgi y en consecuencia, la activación y maduración de las proteínas SREBPs es inhibida (83,84). Una vez en el núcleo, varios cofactores cooperan con las proteínas SREBPs en la transcripción de sus genes blanco, entre ellos se encuentran los factores de transcripción NFY (nuclear transcription factor), Sp1 (specificity protein 1) y CBP (CREB-binding protein) (85). El factor PGC-1β (peroxisome proliferator-activated receptor-γ coactivador-1β) actúa como un coactivador transcripcional de los genes lipogénicos (86).
Regulación de SREBP-1c
Además de poder autorregularse induciendo su propia transcripción (83), SREBP-1c está controlado por factores hormonales y nutritivos, así como por el receptor LXR. La transcripción, el procesamiento proteolítico, la cantidad y la estabilidad de SREBP-1c están controlados por insulina. En hepatocitos aislados de ratón se ha observado que la insulina aumenta hasta 40 veces el ARNm de SREBP-1c (87), mientras que disminuye con el tratamiento con estreptozotocina, un inhibidor de la secreción de insulina (88). La vía de señalización PI3K/AKT-mTORC1 es fundamental en esta regulación, ya que la expresión y la maduración de SREBP-1c se bloquean con rapamicina, un inhibidor de mTORC1 (87). Además, se requiere la activación de la p70S6quinasa (vía PI3K/Akt-mTORC1- p70S6K) (89) para regular el procesamiento proteolítico de SREBP-1c, lo que favorece su maduración. Se ha demostrado además, que la insulina a través de la vía Akt/PKB incrementa la afinidad del complejo SCAP-SREBPs por las proteínas COPII, mientras decrece su afinidad por la proteína Insig, la cual retiene el complejo en la membrana del RE, favoreciendo el procesamiento proteolítico de las proteínas SREBPs (90). La insulina también incrementa los niveles de nSREBPs al inhibir la acción represora de la lipina-1 sobre la función de transcripción de las proteínas SREBPs. Este efecto se verifica mediante la fosforilación de la lipina-1 por mTORC1, impidiendo su depósito en el núcleo, lo que elimina su efecto represor y aumenta la cantidad de nSREBPs (91).
Interacción de insulina y LXR en el control de SREBP-1c
El control transcripcional de SREBP-1c por insulina requiere la presencia del receptor LXR (Figura 2). LXR altamente expresado en el hígado forma heterodímeros (LXR/RXR) con el receptor del ácido 9-cis retinoico (RXR), que en estado basal se une a los elementos de respuesta LXREs en el promotor de sus genes blanco (92). SREBP-1c es un blanco directo de LXR, ya que contiene dos elementos de respuesta a LXR en su promotor (LXRE1, LXRE2), que son fundamentales para la regulación de su transcripción (93), puesto que la insulina, requiere la cooperación de las secuencias LXRE y SRE, para inducir la expresión de SREBP-1(94). De acuerdo con esto, el tratamiento con un agonista de LXR incrementa significativamente la transcripción del gen SREBP-1c, aun en condiciones de una sobrecarga de esterol (95), mientras que el tratamiento con un antagonista de LXR bloquea la activación de la transcripción de SREBP-1 por insulina (94). Sin embargo, el incremento del ARNm de SREBP-1c mediado por LXR parece ser insuficiente para la maduración completa de n-SREBP-1c, lo que se ha atribuido a un procesamiento proteolítico limitado, ya que LXR aumenta la expresión de la proteína Insig-2, con la subsiguiente retención de SREBP-1c en el RE. Por el contrario, en presencia de insulina, que regula a la baja Insig-2, se incrementa el transporte del complejo SREBPs-SCAP al Golgi, aumentando la lipogénesis, lo que ha sugerido que la interacción de insulina y LXR en el control de Insig podría ser un mecanismo protector del exceso de lípidos (96).
Estado nutritivo y SREBP-1c
El estado nutritivo es también un importante regulador de SREBP-1c en hígado, TAB y músculo esquelético. Su expresión disminuye con el ayuno y se incrementa por realimentación con una dieta HCD, a consecuencia del aumento de la glucemia y de la insulinemia (97).Se ha sugerido a este respecto que la glucosa tiene un efecto regulador sobre SREBP-1c mediante una acción directa a nivel transcripcional (98), aunque este concepto está en plena investigación. Sin embargo, se ha demostrado que en respuesta a carbohidratos se requiere la acción sinérgica de ambos factores de transcripción, SREBP-1c y ChREBP, sensible a glucosa, para regular los genes glucolíticos y lipogénicos (99), ya que se ha observado que la deleción de SREBP-1c en el ratón sometido a una dieta HCD, solo induce una disminución del 50% en la síntesis de AG, lo que ha sugerido que la actividad de SREBP-1c no parece ser suficiente, en sí misma, para estimular la total expresión de estos genes (100). Por otra parte, dietas altas en grasa saturada (86) y en fructosa (101) aumentan la respuesta lipogénica en el hígado a través de la activación de SREBP-1c mediada por el coactivador PGC-1β, mientras que por el contrario, los AG PUFA disminuyen la cantidad del nSREBP-1c maduro al inhibir la proteólisis del factor SREBP-1c, a través de la regulación del catabolismo proteasómico de Insig, (102).
Otros reguladores de SREBP-1c
Se han descubierto recientemente otros reguladores, positivos y negativos, de SREBP-1c. En el ratón ob/ob insulin resistente se ha observado que el estrés del RE puede activar SREBP-1c, al eliminar la acción inhibidora de la proteína Insig1 sobre la migración al Golgi de SREBP-1c, aumentando así, su forma madura (nSREBP-1c) (103). También el aminoácido glutamina y la proteína RBP4 activan SREBP-1c al aumentar su expresión y procesamiento a través del Golgi, el primero, (104) e induciendo su maduración y migración al núcleo, la segunda (105). Por el contrario, el factor de crecimiento de fibroblastos 21 (FGF21), la desacetilasa NAD+-dependiente (SIRT1) y la quinasa AMPK regulan a la baja la transcripción, el procesamiento, el transporte al núcleo y la estimulación de SREBP-1c sobre sus genes blanco (106, 107).
El factor SREBP-1c y el HGNA
El factor SREBP-1c hepático juega un papel esencial en el desarrollo del HGNA. El aumento de la concentración del nSREBP-1c es un mecanismo potencial para aumentar la síntesis de novo de los AG en el hígado, al regular al alza los genes glucolíticos (GCK) y lipogénicos (ACLY, ACACA, FASN, GPAT, ELOVL6, SCD1 y LPIN1) en cooperación con LXR y ChREBP, en situaciones de hiperinsulinemia e hiperglucemia (108), como se ha observado en varios modelos animales de RI y en clínica humana (Figura 2). Así, la esteatosis hepática desarrollada en ratones que sobreexpresan SREBP-1c (108) y en ratones ob/ob leptina-deficientes (88), presenta dicho origen. De igual modo, en pacientes con VIH que sufren lipodistrofia, la sobreexpresión hepática de SREBP-1c, se ha asociado con el desarrollo de esteatosis (109), mientras que en pacientes obesos se ha encontrado el único polimorfismo del gen SREBP-1c, que predispone al hígado graso (110). Además, varios estudios experimentales han mostrado el papel fundamental que tiene SREBP-1c como inductor del HGNA, a consecuencia de la ingesta de un exceso de calorías procedentes de grasa (111) o de carbohidratos (101), mientras que por el contrario, la deficiencia hepática de SREBP-1c o la administración de PUFA reducen muy significativamente la infiltración grasa del hígado (100, 112). Por otra parte, la influencia que la alteración de las vías de activación y regulación de SREBP-1c tienen en el desarrollo del HGNA se ha puesto de manifiesto en varios modelos experimentales. Así, un efecto protector sobre la esteatosis desarrollada en el ratón ob/ob o la inducida por sobrealimentación se ha observado, respectivamente, por la deficiencia hepatoespecífica de la Akt2 (113), o por la de raptor (componente del complejo mTORC1), que elimina aparentemente toda la actividad de mTORC1 (91). En contraste, la deleción de TSC1 (tuberous sclerosis protein complex), que funciona como un regulador negativo de TORC1 en la vía Akt, da lugar al aumento de la actividad de TORC1, lo que parece proteger de la esteatosis inducida por la dieta HFD (114). Los autores atribuyen este efecto a la activación de una vía dependiente de Akt, pero resistente a rapamicina, que podría limitar la señal de insulina sobre SREBP-1c y por lo tanto disminuir la síntesis de AG. Asimismo, la deleción de la proteína SCAP parece reducir la lipogénesis y proteger de la esteatosis hepática aún en condiciones de obesidad, hiperinsulinemia e hiperglucemia (115), debido a la disminución del proceso de migración y maduración de SREBP-1c.
Como hemos visto, la inhibición de la vía SREBP-1c y su efecto reductor de la síntesis lipídica puede disminuir el riesgo de la aparición de enfermedades metabólicas como el hígado graso. Por ello, la posibilidad de inhibir diferentes pasos de la vía SREBP-1c, se contempla hoy como una nueva estrategia en el tratamiento de las enfermedades metabólicas en general y del HGNA en particular. El estudio de nuevas moléculas se ha intensificado en los últimos años y, en este sentido, Tang y col (116) encuentran una molécula, la betulina, abundante en la corteza del abedul, que inhibe de forma específica la maduración de SREBP-1c al inducir la interacción entre las proteínas SCAP e Insig y favorecer, por lo tanto, la retención del complejo SCAP-SREBP-1c en el RE. La betulina, además, disminuye la dislipemia y la RI en un modelo de síndrome metabólico, lo que podría ser un prometedor camino en la terapéutica de las alteraciones del metabolismo lipídico (117).
ChREBP
El factor de transcripción ChREBP, también conocido como MONDO A o MLXIPL, fue identificado en 2001 por el grupo de Uyeda (118) en extractos nucleares de tejido hepático de ratas alimentadas con una dieta HCD, como un factor de transcripción que induce la expresión del gen que codifica a la enzima piruvato-quinasa (L-PK), en respuesta a altas concentraciones de glucosa. La glucosa actúa como una molécula señal y, en sinergia con insulina, es necesaria para la inducción de la lipogénesis de novo, como se ha demostrado en el TAB (119) y en el hígado (120), para lo que se requiere su metabolización vía GK (99). En estos tejidos la glucosa estimula la transcripción de genes glucolíticos y lipogénicos, mediante la activación del factor ChREBP y su unión específica al elemento de respuesta a carbohidratos (ChoRE), compuesto por dos cajas E separadas por 5 nucleótidos (5´-CACGTGnnnnnCACGTG-3´) (121). Otras funciones de ChREBP recientemente descubiertas son el control del ritmo circadiano, de la señal redox y del sistema endocrino (122).
ChREBP es un FT de la familia bHLH-LZ que presenta dos isoformas, α y ß. Se ha sugerido que el gen ChREBPα es el primero que se transcribe y este a su vez, activaría posteriormente la transcripción de ChREBPß, mucho más potente (123). ChREBPα es una proteína de 94,6 kDa cuya estructura está muy conservada entre las especies, con una homología de secuencia del 82% entre el hombre, la rata y el ratón (124). La proteína presenta varios dominios: una señal de localización nuclear (NLS), una señal de exportación nuclear (NES), los dominios bHLH-LZ y LZ y dominios de poliprolina. Además, contiene un modulo sensible a glucosa (GSM), que a su vez, consta de un dominio inhibidor a baja glucosa (LID) y un elemento conservado de activación en respuesta a glucosa (GRACE). LID, a su vez, presenta la región conservada MONDO (MCR) I-IV, y GRACE contiene MCR V (122). Por su parte, la isoforma ChREBPß solo contiene GRACE en el módulo GSM (123). La expresión de ambas isoformas de ChREBP es ubicua, aunque muy abundante en hígado, TAB y tejido adiposo marrón (TAM), pero difiere en su localización intracelular. ChREBPα se sitúa en el núcleo y en el citosol celular, mientras ChREBPß presenta localización nuclear. En respuesta a altas concentraciones de glucosa, ChREBPα es rápidamente relocalizado desde el citosol hacia el núcleo (125). Una vez en el núcleo ChREBP debe unirse al elemento de respuesta a carbohidratos (ChoRE), en la región promotora de los genes glucolíticos y lipogénicos, lo que requiere la heterodimerización de ChREBP con la proteína Mlx (Max-like protein), un miembro de la familia de FT (bHLH-LZ), Myc/Max/Mad, que es un cofactor obligado de ChREBP en la regulación de estos genes hepáticos. Se configura así el complejo ChREBP/Mlx como el principal mediador de la expresión génica en respuesta a glucosa (126).
Regulación de la actividad de ChREBP
Se han postulado varias modificaciones postraduccionales que regulan la actividad de ChREBP en respuesta a glucosa. El mecanismo de fosforilación/defosforilación, el mejor caracterizado, puede actuar como un regulador positivo o negativo de ChREBP y tiene gran importancia para su localización intracelular. Así, la proteína-quinasa activada por AMPc (PKA) y la AMPK, regulan negativamente ChREBP, mediante la fosforilación de los residuos Ser196, Ser626 y Treo666 (PKA) y de Ser568 (AMPK) de su molécula, reteniéndolo en el citosol en respuesta a bajas concentraciones de glucosa, a dietas HFD o en situaciones de ayuno (127), mientras que por el contrario, la defosforilación de ChREBP en el residuo Ser196, induce su entrada en el núcleo en respuesta a la sobrecarga de glucosa (128). Otras posibles modificaciones postraduccionales de ChREBP son la acetilación en el residuo Lys 67, por acción del coactivador p300 con actividad histona acetiltransferasa (HAT) (129) y la O-glicosilación por activación de la O-N-acetilglucosamina-transferasa (OGT), enzima que transfiere el monosacárido N-acetilglucosamina a los residuos Ser/Treo de ChREBP (130). Ambas son reguladores positivos del factor, aumentando sus niveles y su capacidad de transcripción. En este sentido se ha observado que el aumento de la acetilación y la o-glicosilación de ChREBP, bajo condiciones de hiperglucemia (ratón ob/ob y db/db), incrementan su activación, favoreciendo el desarrollo de esteatosis hepática (129, 131).
Metabolitos implicados en la activación del complejo ChREBP/Mondo-Mlx.
Varios metabolitos de la glucosa han sido implicados en la activación de ChREBP/Mondo-Mlx. Se ha señalado a la enzima xilulosa 5-fosfato (Xu-5P), un intermediario de la vía de las pentosas fosfato, como un posible mediador que induce la entrada de ChREBP en el núcleo. El mecanismo implica la activación de la fosfatasa 2A (PP2A), que defosforila ChREBP en el residuo Ser196. En el núcleo una segunda defosforilación en el residuo Treo666, PP2A-inducida, le permite al factor ChREBP unirse a las secuencias ChoRE de los genes blanco, induciendo su transcripción (132). Otros metabolitos (glucosa-6-fosfato (Glu-6-P) y fructosa 2,6-bisfosfato (F-2,6P2)) también se han propuesto como activadores de ChREBP. Los trabajos de Dentin y col (133), señalan al metabolito Glu-6-P, como la molécula fundamental de activación de ChREBP, ya que demuestran que la sobreexpresión de la enzima glucosa-6-fosfato deshidrogenasa (G6-PDH) suprime la actividad de ChREBP a través de la disminución de los niveles de Glu-6-P y, viceversa, la deleción de G6-PDH incrementa la actividad transcripcional de ChREBP por aumento de los niveles de Glu-6-P. Por otra parte, el metabolito F-2,6P2 que se sintetiza a partir de fructosa-6-fosfato (F-6-P) y es degradado de nuevo a F-6-P por la acción de la enzima bifuncional fructosa 6-fosfofructo-2-quinasa/fructosa-2.6-bisfosfatasa (PFK-2/FBPasa2), ha sido implicado en la respuesta de ChREBP a glucosa en hepatocitos. Se ha comprobado a este respecto que la depleción selectiva de F-2,6P2, por una variante deficiente de la actividad bisfosfatasa de PFK-2/FBPasa2, inhibe la unión de ChREBP a sus genes blanco (134).
Regulación transcripcional de ChREBP
Los mecanismos moleculares que subyacen en la regulación transcripcional del gen ChREBP no son bien conocidos. Se ha sugerido que ChREBP puede regular su propia expresión en respuesta a glucosa (126), pero estudios recientes han mostrado que LXR y el receptor de la hormona tiroidea ß (TR-ß) son los reguladores trascripcionales básicos de este factor en el hígado, por su unión en la región promotora del gen a los elementos de respuesta, LXRE1 y LXRE2 (Figura 2) (135,136). El heterodímero LXR/RXR presenta una gran afinidad por LXRE1, regulando al alza la transcripción de ChREBP como gen diana directo de LXRα (135). Cha y Repa (135) encuentran que el tratamiento con agonistas de LXR y RXRs aumenta hasta 6 veces el ARNm de ChREBP en el hígado del ratón salvaje, pero no en el doble knockout de LXR (LXRαß-/-). Además, el hecho de que la deficiencia en LXR disminuya hasta un 80% la lipogénesis hepática, un porcentaje superior al producido por la deleción independiente de SREBP-1c (50%) (100) y ChREBP (60%) (137,138)), señala la dependencia directa de ambos respecto a la estimulación de LXR para ejercer sus funciones. El receptor nuclear TR-ß, por su parte, previa dimerización con RXR (TR-ß/RXR), también regula positivamente la expresión génica de ChREBP por su unión específica a la secuencia LXRE2, pero no a LXRE1 (que resulta ser preferente para LXR), aunque se ha demostrado, que la inducción de la expresión de ChREBP por TR-ß, es independiente de LXR (136). Por último, la insulina es otro regulador positivo de ChREBP que actúa mediante la disminución de octamer-1 (Oct1), un represor de la transcripción que se une a su promotor (139).
ChREBP, lipogénesis, RI e HGNA
ChREBP, como intermediario de LXR y actuando en sinergia con SREBP-1c, regula al alza la expresión de genes implicados en el metabolismo de glúcidos y lípidos, incrementando la lipogénesis de novo (Figura 2). Usando un dominante negativo de Mlx (dnMlx) se han analizado los genes blanco cuya expresión está regulada por ChREBP/Mlx. Entre ellos destacan genes lipogénicos (ACACA, FASN, ELOVL6, SCD1 Y GPAT), glucolíticos (PKLR), gluconeogénicos (GLPC (glu-6-Pasa, subunidad catalítica)) y PEPCK (fosfoenolpiruvato-carboxiquinasa), de la vía de las pentosas fosfato (G6PD (G6-PDH) y TKT (transcetolasa) y el gen S14 del enzima Spot 14, inducible por glucosa, que presenta una acción específica de tejido promotora de la lipogénesis (126, 140). De acuerdo con este perfil funcional, el factor ChREBP puede considerarse como un mediador de la conversión de un exceso de carbohidratos en lípidos, cuya activación, puede inducir el desarrollo del HGNA. Este efecto proesteatótico de la activación de ChREBP se ha confirmado en el hígado de pacientes con EHNA y en sujetos obesos (141, 142), en los que se observa un aumento de la expresión del ARNm de ChREBP, mientras que por el contrario, en el ratón knockout (ChREBP-/-) se reducen las vías glucolítica y lipogénica hepáticas y los AGL circulantes (137). Sin embargo, los datos actuales del papel jugado por ChREBP en el metabolismo de la glucosa y en especial sobre la RI y su relación con la lipogénesis durante el desarrollo del HGNA, son confusos y contradictorios. Así, mientras la deficiencia global de ChREBP, reduce la lipogénesis y mantiene la RI (137), su inhibición hepatoespecífica o la sobreexpresión de un antagonista (dnMlx) mejora ambas, la esteatosis y la RI, en el hígado del ratón ob/ob (143) y en el ratón C57BL/6J diabético (144), lo que se ha asociado con un aumento de la actividad de la vía AKT/PKB y de las cinasas ERK1 y ERK2 hepáticas (145). Teniendo en cuenta los dos fenotipos, Benhamed y col (142) han propuesto que este efecto beneficioso de la deficiencia de ChREBP sobre el metabolismo de la glucosa, aumentando la sensibilidad a la insulina, solo puede producirse en el caso de una sobrecarga de lípidos. El trabajo realizado por este grupo, que puede cambiar el concepto actual del papel que ChREBP tiene en la esteatosis hepática y en la RI, muestra que la sobreexpresión de ChREBP en ratones sometidos a una dieta estándar desarrolla esteatosis hepática, pero permanecen insulin-sensibles. Más interesante aún, ratones alimentados con una dieta HFD y que sobreexpresan ChREBP presentan una insulinemia normal y una mejora de la señalización insulínica y de la tolerancia a la glucosa, a pesar de desarrollar una esteatosis hepática masiva en relación al control. Los autores atribuyen la mejora de la sensibilidad insulínica a la modificación de las especies lipídicas que componen el depósito graso del hígado y que resulta del incremento de los AG monoinsaturados (MUFAs), respecto a los AGS lipotóxicos como el palmitato, que disminuyen, modificando el balance MUFA/AGS a favor de MUFA. El aumento de la expresión de la enzima SCD1, cuyo gen es blanco de ChREBP y que convierte los AGS en MUFA, parece ser fundamental para implementar el efecto beneficioso que la activación de ChREBP tiene en la esteatosis hepática. Benhamed y col (142) postulan que la estimulación de ChREBP parece disociar la esteatosis hepática de la RI, generando efectos paliativos sobre ambos, el metabolismo glucídico y el lipídico, al tamponar los AG lipotóxicos y favorecer la partición de los lípidos en el tejido. Este resultado refuerza el concepto de que no todos los lípidos son perjudiciales para la sensibilidad a la insulina y que, almacenados en el espacio y en el tiempo en zonas adecuadas, pueden generar señales que modulen la adaptación al estrés (146). Entre las consecuencias que se pueden derivar de estos resultados, se encuentran en primer lugar, la reconsideración del concepto del factor ChREBP como promotor de la esteatosis hepática y de la RI y en segundo, la posibilidad de explorar la expresión de ChREBP como un posible blanco terapéutico del HGNA y de otras manifestaciones del síndrome metabólico (147).
RECEPTOR X HEPÁTICO (LXR)
Los receptores LXR son factores de transcripción activados por ligando que pertenecen a la superfamilia de receptores nucleares (RN) (148) y cuya función es esencial en la regulación del metabolismo del COL y de los ácidos biliares y en el control de la homeostasis lipídica y glucídica y de la inflamación, en respuesta a oxiesteroles (138, 149, 150). Existen dos isoformas de LXR (LXRα y LXRß), que presentan entre sí un 78% de identidad y que son codificadas respectivamente, por los genes NR1H3 y NR1H2 (151). LXRα se expresa mayoritariamente en el hígado y en menor medida en intestino, TAB, riñón y macrófagos y su función primordial es regular de forma específica de tejido, el metabolismo del COL y la lipogénesis de novo, como se ha observado en el ratón deficiente en LXRα (LXRα-/-) y en el doble deficiente (LXRαß-/-) (138, 149, 152). La isoforma LXRß, por su parte, tiene una expresión ubicua y actúa de forma primordial como un regulador del metabolismo glucídico y de la homeostasis energética en el TAB y en músculo estriado (153). A este respecto, en el ratón LXRß-/-, se ha observado un aumento del gasto energético y un efecto protector frente a la obesidad inducida por dieta, que se acompaña de un aumento de la expresión de la proteína desacoplante 1 (UPC1) en varios tejidos, en especial en el TAM (153,154).
Su estructura, común para todos los RN, presenta un dominio activador funcional de la transcripción (AF-1) N-terminal, independiente de ligando; un dominio de interacción al ADN (DBD), que contiene dos dedos de zinc que interaccionan con los sitios DR4 en el promotor de los genes blanco; un dominio D, donde se produce la interacción con cofactores y un dominio de unión a ligandos C-terminal (LBD), que contiene una función activadora dependiente de ligando (AF-2), que media la activación de la maquinaria de transcripción (155).
Activación de LXR
Según el mecanismo de activación convencional, el heterodímero (LXR/RXR) en ausencia de ligando se une al ADN sobre sus elementos de respuesta (LXREs), compuestos por dos secuencias (5´-AGGTCA-3´), separadas por cuatro nucleótidos (DR4)(93) e interacciona con corepresores, como NCoR (nuclear receptor co-represor) o SMRT (silencing mediator of retinoid and thyroid receptors), que bloquean la transcripción al unirse con proteínas con actividad histona desacetilasa (HDAC), a través de la proteína Sin3A (stress-activated MAPkinase interacting proteín 3)(156). Una vez unido al ligando, LXR cambia su conformación, liberando los corepresores y reclutando coactivadores como ASC2 (activating signal cointegrator-2) o RIP140 (receptor interacting protein 140) sobre el LBD, que permite a la cromatina tener un estado permisivo para iniciar la transcripción (157,158).
Además de activarse también por un mecanismo de activación alternativa, mediante la modulación por marcadores epigenéticos (159), LXR puede regular negativamente la expresión de genes inflamatorios que contienen sitios NF-kB, AP-1 o STATs (signal transducer and activator of transcription), por el mecanismo de transrepresión dependiente de ligando (160).
Regulación de LXR
La activación de LXR y su unión al ADN está regulada por una interacción compleja entre sus ligandos, los cofactores, las hormonas y las modificaciones postraduccionales que le afectan. Varios tipos de oxiesteroles, entre ellos, 20(S)-hidroxicolesterol, 22(R) hidroxicolesterol, 24(S)-hidroxicolesterol, 25-hidroxicolesterol, 27-hidroxicolesterol y 24(S) y 25-epoxicolesterol, derivados de la oxidación del COL, son ligandos naturales activadores de LXR en hepatocitos y en otros tipos celulares (161). También se han utilizado, tanto in vivo como in vitro (162), agonistas sintéticos de LXR como T091317, GW3965 o WYE-672, mientras que los AG PUFA actúan como antagonistas (163). Por otra parte, los corepresores NCoR y SMRT (156) son reguladores negativos de LXR, mientras el cofactor PPARγ coactivador-1α (PGC-1α), parece potenciar su actividad (164).
Entre las modificaciones postraduccionales que pueden afectar a LXR, podemos destacar la desacetilación por SIRT-1, en respuesta a la disponibilidad de nutrientes. El cambio de conformación de LXR, una vez unido a su ligando, favorece su interacción con SIRT1 en residuos de lisina, desencadenando la ubiquitinación y degradación del receptor, mecanismo importante para su reciclaje y activación (165). Además, la fosforilación de LXR por PKA, que regula negativamente la actividad de LXR, impide la transcripción de SREBP-1c en los hepatocitos (166). Otra importante modificación de LXR es la sumoilación, que resulta fundamental para la transactivación de los genes antiinflamatorios (160).
Por otra parte, en estudios in vitro e in vivo, se ha observado que la insulina estimula al alza la expresión del ARNm de LXRα en hepatocitos, mientras que este disminuye, junto a la de otros genes lipogénicos (ACC, FAS), en los ratones knockout LXRα-/- y LXRαß-/- tratados con la hormona (167), lo que confirma el papel fundamental de LXR como sensor de la insulina en la estimulación fisiológica de la expresión de SREBP-1 (94). El mecanismo propuesto sugiere que la insulina inhibiría por fosforilación mediante la proteína quinasa B (PKB), la forma activa de FoxO1, eliminando su efecto inhibidor sobre la unión de LXR a los LXREs del promotor de SREBP-1c, favoreciendo su transcripción (168). Por último, los RN PPARα y PPARγ son reguladores positivos de la expresión de LXR (169), mientras el receptor X farnesoide (FXR), un FT que es activado por los ácidos biliares, reprime la actividad de LXR mediante la inducción de la proteína SHP (small heterodimer partner), un RN huérfano, atípico, que no contiene el dominio DBD (170).
LXR y el metabolismo del colesterol
LXR fue inicialmente identificado por su función de sensor nuclear del COL (138, 149). Su función, esencial en el metabolismo y en el aclaramiento del COL en respuesta a oxiesteroles y a agonistas sintéticos, contribuye a la eliminación del exceso de COL en el organismo. Estudios en modelos trangénicos de ratón han puesto de manifiesto que LXR induce la síntesis de ácidos biliares a partir del COL hepático, regulando al alza el gen CYP7A1 que codifica a la colesterol-7α-hidroxilasa, enzima limitante de dicha síntesis (149), mientras que por el contrario, el hígado del ratón LXRα-/- alimentado con una dieta alta en COL, presenta una sobrecarga de COL esterificado (171). La administración in vivo de agonistas de LXR también incrementa el aclaramiento del COL mediante la regulación al alza de la expresión de genes relacionados con la absorción, la excreción y el transporte reverso del COL (RCT), como los transportadores ABCA1, ABCG1/ABCG4 y ABCG5/ABCG8 en hígado, intestino y macrófagos (172). En este sentido, el hecho de que la inducción de la proteína ABCA1 intestinal por activación de LXRα eleve las lipoproteínas HDL en plasma, parece indicar que el efecto sobre el RTC se produce fuera del hígado y sugiere que el LXR intestinal tiene importancia en la biogénesis del HDL-colesterol, vía ABCA1 (173). Además, el aumento de la expresión de la apolipoproteína E asociada a HDL, que facilita el flujo del COL celular, ha sugerido un papel protector de LXR en la aterosclerosis (174).
LXR y el metabolismo de la glucosa
En modelos murinos de RI se ha demostrado que agonistas sintéticos de LXR incrementan la sensibilidad a la insulina y reducen la glucemia (175, 176). El efecto neto sería el aumento de la utilización de la glucosa y la reducción de su producción hepática, aunque los datos existentes son contradictorios. Una menor producción de glucosa hepática se ha observado en el ratón db/db (175) y en ratas alimentadas con una dieta HFD (177) por activación de LXR, debido a la regulación a la baja de la expresión génica de las enzimas gluconeogénicas PEPCK y Glu-6-Pasa. Por otra parte, el efecto sensibilizador a la insulina de LXR se asocia con el aumento de la captación de la glucosa basal y la estimulada por insulina, a través de la regulación al alza de los transportadores GLUT4 y GLUT1, observado en adipocitos 3T3-L1 que sobreexpresan LXR (176) y en el TAB de ratones tratados con el agonista GW3965 (178). Este efecto beneficioso sobre el metabolismo de la glucosa parece mediado por ambas isoformas de LXR, aunque solo el ratón LXRα-/-, muestra un nivel disminuido de la expresión de GLUT4 (179). Por el contrario, otros estudios o no han podido reproducir estos efectos (180) u observan un efecto negativo de LXR, sobre la captación de glucosa. En este sentido, Pettersson y col. (2013) (181) señalan que la activación de LXR en adipocitos humanos inhibe la traslocación a la membrana de GLUT4 y suprime varios genes que codifican mediadores de la señalización insulínica (Akt2, la proteína asociada a c-Cbl (CAP) y la caveolina1), generando un efecto diabetógeno. Además, aunque ambas isoformas LXRα y LXRß inducen la secreción de insulina estimulada por glucosa (182), este efecto se contrarresta por el aumento de la lipogénesis pancreática derivada de la activación de LXR, que conduce a lipotoxicidad, daño celular y disfunción de la célula beta (183).
LXRα. lipogénesis e HGNA
El receptor LXR es el regulador fundamental de la biosíntesis de los AG en el hígado, función que lleva a cabo mediante un doble mecanismo: un efecto directo sobre los genes que codifican las enzimas lipogénicas ACC, FAS y SCD-1, por interacción con los LXREs funcionales contenidos en su promotor (184, 185,186) y otro indirecto, ya señalado, que se efectúa mediante el control de la expresión génica de SREBP-1c y CHREBP, que son dianas de LXR (Figura 2) (94, 135,186). Por ello, la sobreexpresión de LXR en un modelo diabético de ratón (db/db) (187) o la administración del agonista T0901317 (188), inducen una esteatosis hepática masiva, trigliceridemia y un incremento de la secreción de VLDL. En sentido opuesto, ratones knockout (LXRα-/-) muestran una disminución en la expresión de los genes lipogénicos indicados (149), lo que confirma su implicación en el desarrollo del HGNA. El mediador primordial del efecto prolipogénico de LXR es la isoforma LXRα (138,149), aunque recientemente se ha demostrado la participación de LXRß en el proceso, ya que el efecto estimulador del agonista T0901317 sobre la lipogénesis de novo, se reduce en el ratón LXRß-/- (189) y además este subtipo parece mediar la esteatosis hepática inducida por glucocorticoides (190).
Por lo tanto, LXR como regulador principal junto con SREBP-1c y CHREBP forman una red de FT que, de forma coordinada, controlan la lipogénesis de novo hepática y cuya sobreexpresión media la infiltración grasa del hígado en respuesta a la hiperinsulinemia y la hiperglucemia desarrolladas en estados de RI, como en el HGNA (Figura 2)(191, 192).
LXR e inflamación
Estudios recientes hacen énfasis en el papel fundamental que LXR parece tener en la integración de las señales metabólica e inflamatoria (193) y su proyección en numerosas enfermedades, entre ellas las relacionadas con el síndrome metabólico, en las que estos aspectos están intrínsecamente unidos (194). A este respecto, se han identificado a las isoformas LXRα y LXRß como factores de transcripción antiinflamatorios y reguladores fisiológicos de la fagocitosis, de la respuesta inmune y de la apoptosis (195). Mediante el mecanismo de transrepresión, LXR regula a la baja la expresión de genes inflamatorios y de la inmunidad innata en diversos tipos celulares, incluyendo los hepatocitos (160), las células de Kupffer (196) y las células estrelladas (HSC) del hígado (197).Los primeros datos al respecto indican que la activación de los LXRs en macrófagos, antagoniza la expresión de iNOS y COX-2, en respuesta a la estimulación con endotoxina (LPS), TNFα o IL-1ß, mediante la inactivación de los factores de transcripción NF-kB, STATs y AP-1 (193). Del mismo modo, el tratamiento de las células de Kupffer con el agonista GW3965 interfiere con la regulación postranscripcional de la producción de TNFα y la activación de la quinasa MAPK p38, inducidos por LPS, reduciendo los niveles de TNFα y protegiendo al hígado de la endotoxemia (196). Asimismo, ambas isoformas, LXRα y LXRß, inhiben la expresión de las proteínas de fase aguda, aunque LXRß parece ser el mediador principal (160). En otro sentido, un efecto inhibidor de LXR sobre la activación de las células HSC hepáticas se ha descrito recientemente (198). En este estudio la estimulación de LXR mantiene el fenotipo quiescente de las células HSC, disminuyendo la producción de marcadores de la fibrogénesis, mientras por el contrario, la doble deficiencia de LXR (LXRαβ-/-), aumenta su capacidad fibrogénica, haciendo al hígado más susceptible al trauma. Todos estos datos han sugerido que la estimulación de LXR podría ser beneficiosa en la progresión del HGNA a la EHNA, no solo por su efecto preventivo de la alteración de las células de Kupffer y de las HSC (196,197), sino también por su capacidad de aumentar la excreción del COL libre, un lípido que actúa como una molécula lipotóxica, capaz de inducir la inflamación (199, 200).
Debido a sus funciones antiaterogénica, antidiabética y antiinflamatoria, en los últimos años se ha intentado el uso en terapéutica de LXR y de sus agonistas, pero su utilización se ha visto limitada por el efecto deletéreo que genera sobre el metabolismo lipídico desarrollando hígado graso. La búsqueda de nuevos agonistas de LXR que manteniendo sus efectos beneficiosos puedan reducir al mismo tiempo los prolipogénicos, ha sido intensa, aunque en principio la aparición de otros efectos colaterales en algunos compuestos, ha conducido a abandonar su uso. Actualmente sin embargo, la investigación se orienta en la síntesis de moléculas que puedan disecar las funciones individuales y muy específicas de los subtipos de LXR, obviando así, los efectos indeseados (201). En relación a un posible tratamiento de la esteatosis y de otras enfermedades metabólicas, muy recientemente se ha descrito un agonista inverso de LXR (SR9238), hepato-selectivo, que suprime además de la colesterolemia, la lipogénesis y el daño hepáticos en un modelo experimental de obesidad inducida por dieta HFD, lo que abre un camino muy relevante en la prevención del HGNA y quizá de su progresión a la EHNA (202). Sin embargo, para cumplir con el objetivo de obtener moléculas beneficiosas en el tratamiento del HGNA y EHNA, la investigación actual se orienta hacia el estudio de la interacción funcional entre LXR, SREBP-1c y CHREBP con otros reguladores del metabolismo de glúcidos y de lípidos tales como el receptor FXR, el receptor X pregnano (PXR), el receptor constitutivo de androstano (CAR), el receptor de la vitamina D (VDR) y los receptores de la familia PPARs (α β y γ), a fin de poder conocer en toda su dimensión, la regulación molecular de la vía lipogénica y su proyección en la patogenia de la esteatosis y su progresión a la EHNA, abriendo la posibilidad de encontrar nuevas dianas, útiles en la terapia de esta enfermedad (194) (203)(204).
CONCLUSIÓN
En los últimos años se ha progresado de forma sustancial en la comprensión de los mecanismos moleculares y celulares que subyacen en la patogenia del HGNA y su progresión hacia la EHNA. El HGNA puede ser considerado como la manifestación hepática del síndrome metabólico cuya primera causa es la RI. El acúmulo de TG en los hepatocitos es básicamente el resultado del aumento del aporte de AGL circulantes y de la lipogénesis de novo. Los factores de transcripción SREBP-1c, ChREBP y LXR, forman una red funcional interactiva que regula al alza la transcripción de genes que codifican enzimas de la biosíntesis de los AG y de los TG en respuesta a insulina, glucosa y oxiesteroles y que están implicados en el desarrollo del HGNA. Los datos actuales permiten dirigir la atención en varias direcciones: el estudio del papel diferencial y específico que el factor ChREBPβ parece tener en el hígado respecto al TAB, en donde se postula un efecto reductor de la lipogénesis de novo y de la RI en la obesidad humana (205); un mayor conocimiento de la interrelación funcional de todos los FT implicados en la regulación de los factores que afectan al HGNA; y el desarrollo de agonistas selectivos, sin efectos secundarios, que puedan tener efectos terapéuticos específicos sobre el HGNA y su progresión a la EHNA.
REFERENCIAS
1. Farrell, G.C., Larter, C.Z., 2006. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43, S99-S112.
2. Angulo, P., 2002. Nonalcoholic fatty liver disease. N Engl J Med 346, 1221-1231.
3. Ludwig, J., Viggiano, T.R., McGill, D.B., Oh, B.J., 1980. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 55, 434-438.
4. Kopec, K.L., Burns, D., 2011. Nonalcoholic fatty liver disease: a review of the spectrum of disease, diagnosis, and therapy. Nutr Clin Pract 26, 565-576.
5. Adams, L.A., Lindor, K.D., 2007. Nonalcoholic fatty liver disease. Ann Epidemiol 17, 863-869.
6. Marchesini, G., Babini, M., 2006. Nonalcoholic fatty liver disease and the metabolic syndrome. Minerva Cardioangiol 54, 229-239.
7. Edens, M.A., Kuipers, F., Stolk, R.P., 2009. Non-alcoholic fatty liver disease is associated with cardiovascular disease risk markers. Obes Rev 10, 412-419.
8. Caballería, L., Pera, G., Auladell, M.A. y col. 2010. Prevalence and factors associated with the presence of nonalcoholic fatty liver disease in an adult population in Spain. Eur J Gastroenterol Hepatol 22, 24-32.
9. Bellentani, S., Scaglioni, F., Marino, M., Bedogni, G., 2010. Epidemiology of non-alcoholic fatty liver disease. Dig Dis 28, 155-161.
10. Baumeister, S.E., Völzke, H., Marschall, P. y col. 2008. Impact of fatty liver disease on health care utilization and costs in a general population: a 5-year observation. Gastroenterology 134, 85-94.
11. Calori, G., Lattuada, G., Ragogna, F. y col. 2011. Fatty liver index and mortality: the Cremona study in the 15th year of follow-up. Hepatology 54, 145-152.
12. Kanuri, G., Bergheim, I., 2013. In Vitro and in Vivo Models of Non-Alcoholic Fatty Liver Disease (NAFLD). Int J Mol Sci 14, 11963-11980.
13. Bugianesi, E., McCullough, A.J., Marchesini, G., 2005. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 42, 987-1000.
14. Morino, K., Petersen, K.F., Shulman, G.I., 2006. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 55 Suppl 2, S9-S15.
15. Kotronen, A., Yki-Järvinen, H., 2008. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol 28, 27-38.
16. Savage, D.B., 2009. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest 119, 315-322.
17. Gonzalez-Baró, M.R., Lewin, T.M., Coleman, R.A., 2007. Regulation of Triglyceride Metabolism. II. Function of mitochondrial GPAT1 in the regulation of triacylglycerol biosynthesis and insulin action. Am J Physiol Gastrointest Liver Physiol 292, G1195-1199.
18. Yamaguchi, K., Yang, L., McCall, S. y col. 2007. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45, 1366-1374.
19. Monetti, M., Levin, M.C., Watt, M.J. y col. 2007. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 6, 69-78.
20. Minehira, K., Young, S.G., Villanueva, C.J. y col. 2008. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res 49, 2038-2044.
21. Samuel, V.T., Liu, Z.X., Qu, X. y col. 2004. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 279, 32345-32353.
22. Samuel, V.T. y Shulman G.I. 2012. Integrating mechanisms for insulin resistance. Common threads and missing links. Cell 148. 852-871.
23. Donnelly, K.L., Smith, C.I., Schwarzenberg, S.J. y col. 2005. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115, 1343-1351.
24. Ginsberg, H.N., Zhang, Y.L., Hernandez-Ono, A., 2005. Regulation of plasma triglycerides in insulin resistance and diabetes. Arch Med Res 36, 232-240.
25. Greco, D., Kotronen, A., Westerbacka, J. y col. 2008. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol 294, G1281-1287.
26. Hubbard, B., Doege, H., Punreddy, S. y col. 2006. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity. Gastroenterology 130, 1259-1269.
27. Fabbrini, E., Sullivan, S., Klein, S., 2010. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 51, 679-689.
28. Foufelle, F., Ferré, P., 2002. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c.
29. Munday, M.R., 2002. Regulation of mammalian acetyl-CoA carboxylase. Biochem Soc Trans 30, 1059-1064.
30. Smith, S., Witkowski, A., Joshi, A.K., 2003. Structural and functional organization of the animal fatty acid synthase. Prog Lipid Res 42, 289-317.
31. Ntambi, J.M., Miyazaki, M., 2004. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog Lipid Res 43, 91-104.
32. Yen, C.L., Stone, S.J., Koliwad, S. y col. 2008. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 49, 2283-2301.
33. Chen, G., Liang, G., Ou, J. y col. 2004. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci U S A 101, 11245-11250.
34. Shimano, H., 2001. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res 40, 439-452.
35. Dentin, R., Girard, J., Postic, C., 2005. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 87, 81-86.
36. Tessari, P., Coracina, A., Cosma, A., Tiengo, A., 2009. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis 19, 291-302.
37. Kotronen, A., Seppälä-Lindroos, A., Vehkavaara, S. y col. 2009. Liver fat and lipid oxidation in humans. Liver Int 29, 1439-1446.
38. Lavoie, J.M., Gauthier, M.S., 2006. Regulation of fat metabolism in the liver: link to non-alcoholic hepatic steatosis and impact of physical exercise. Cell Mol Life Sci 63, 1393-1409.
39. Reddy, J.K., Rao, M.S., 2006. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol 290, G852-858.
40. Leclercq, I.A., Farrell, G.C., Field, J. y col. 2000. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest 105, 1067-1075.
41. Louet, J.F., Chatelain, F., Decaux, J.F. y col. 2001. Long-chain fatty acids regulate liver carnitine palmitoyltransferase I gene (L-CPT I) expression through a peroxisome-proliferator-activated receptor alpha (PPARalpha)-independent pathway. Biochem J 354, 189-197.
42. Reddy, J.K., Hashimoto, T., 2001. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr 21, 193-230.
43. Akiyama, T.E., Nicol, C.J., Fievet, C. y col. 2001. Peroxisome proliferator-activated receptor-alpha regulates lipid homeostasis, but is not associated with obesity: studies with congenic mouse lines. J Biol Chem 276, 39088-39093.
44. Stienstra, R., Mandard, S., Patsouris, D. y col. 2007. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology 148, 2753-2763.
45. Harano, Y., Yasui, K., Toyama, T. y col. 2006. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int 26, 613-620.
46. Adiels, M., Taskinen, M.R., Packard, C. y col. 2006. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 49, 755-765.
47. Adiels, M., Westerbacka, J., Soro-Paavonen, A. y col. 2007. Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia 50, 2356-2365.
48. Choi, S.H., Ginsberg, H.N., 2011. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab 22, 353-363.
49. Day, C.P., James, O.F., 1998. Steatohepatitis: a tale of two "hits"? Gastroenterology 114, 842-845.
50. Trauner, M., Arrese, M., Wagner, M., 2010. Fatty liver and lipotoxicity. Biochim Biophys Acta 1801, 299-310.
51. Pérez-Carreras, M., Del Hoyo, P., Martín, M.A. y col. 2003. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 38, 999-1007.
52. Garcia-Ruiz, C., Fernandez-Checa, J.C., 2006. Mitochondrial glutathione: hepatocellular survival-death switch. J Gastroenterol Hepatol 21 Suppl 3, S3-6.
53. Maher, J.M., 2006. Mechanisms of liver injury due to fat", AASLD Postgrad. Course Syllabus, p.89-96.
54. Pessayre, D., Fromenty, B., 2005. NASH: a mitochondrial disease. J Hepatol 42, 928-940.
55. Grieco, A., Forgione, A., Miele, L. y col. 2005. Fatty liver and drugs. Eur Rev Med Pharmacol Sci 9, 261-263.
56. Wieckowska, A., Zein, N.N., Yerian, L.M. y col. 2006. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 44, 27-33.
57. Carter-Kent, C., Zein, N.N., Feldstein, A.E., 2008. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: implications for treatment. Am J Gastroenterol 103, 1036-1042.
58. Feldstein, A.E., 2010. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin Liver Dis 30, 391-401.
59. Stienstra, R., Saudale, F., Duval, C. y col. 2010. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 51, 511-522.
60. Leonarduzzi, G., Scavazza, A., Biasi, F. y col. 1997. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor beta1 expression in the macrophage lineage: a link between oxidative injury and fibrosclerosis. FASEB J 11, 851-857.
61. Kaplowitz, N., Than, T.A., Shinohara, M., Ji, C., 2007. Endoplasmic reticulum stress and liver injury. Semin Liver Dis 27, 367-377.
62. Kammoun, H.L., Hainault, I., Ferré, P., Foufelle, F., 2009. Nutritional related liver disease: targeting the endoplasmic reticulum stress. Curr Opin Clin Nutr Metab Care 12, 575-582.
63. Ron, D., Walter, P., 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8, 519-529.
64. Gentile, C.L., Frye, M., Pagliassotti, M.J., 2011. Endoplasmic reticulum stress and the unfolded protein response in nonalcoholic fatty liver disease. Antioxid Redox Signal 15, 505-521.
65. Wang, D., Wei, Y., Pagliassotti, M.J., 2006. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 147, 943-951.
66. Schenk, S., Saberi, M., Olefsky, J.M., 2008. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 118, 2992-3002.
67. Yamauchi, T., Kamon, J., Ito, Y. y col. 2003. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762-769.
68. Yamauchi, T., Kamon, J., Minokoshi, Y. y col. 2002. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8, 1288-1295.
69. Wu, X., Mahadev, K., Fuchsel, L. y col. 2007. Adiponectin suppresses IkappaB kinase activation induced by tumor necrosis factor-alpha or high glucose in endothelial cells: role of cAMP and AMP kinase signaling. Am J Physiol Endocrinol Metab 293, E1836-1844.
70. Xu, A., Wang, Y., Keshaw, H. y col. 2003. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 112, 91-100.
71. Targher, G., Bertolini, L., Scala, L. y col. 2004. Decreased plasma adiponectin concentrations are closely associated with nonalcoholic hepatic steatosis in obese individuals. Clin Endocrinol (Oxf) 61, 700-703.
72. Bertolani, C., Sancho-Bru, P., Failli, P. y col. 2006. Resistin as an intrahepatic cytokine: overexpression during chronic injury and induction of proinflammatory actions in hepatic stellate cells. Am J Pathol 169, 2042-2053.
73. Marra, F., 2007. Leptin and liver tissue repair: do rodent models provide the answers? J Hepatol 46, 12-18.
74. Ota, T., Takamura, T., Kurita, S. y col. 2007. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology 132, 282-293.
75. Fuchs, M., Sanyal, A.J., 2012. Lipotoxicity in NASH. J Hepatol 56, 291-293.
76. Hotta, K., Yoneda, M., Hyogo, H. y col. 2010. Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Med Genet 11, 172.
77. Petta, S., Muratore, C., Craxì, A., 2009. Non-alcoholic fatty liver disease pathogenesis: the present and the future. Dig Liver Dis 41, 615-625.
78. Horton, J.D., Goldstein, J.L., Brown, M.S., 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109, 1125-1131.
79. Hua, X., Wu, J., Goldstein, J.L., Brown, M.S., Hobbs, H.H., 1995. Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics 25, 667-673.
80. Shimomura, I., Shimano, H., Horton, J.D. y col. 1997. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 99, 838-845.
81. Foretz, M., Guichard, C., Ferré, P., Foufelle, F., 1999. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci U S A 96, 12737-12742.
82. Sato, R., 2010. Sterol metabolism and SREBP activation. Arch Biochem Biophys 501, 177-181.
83. Daemen, S., Kutmon, M., Evelo, C.T., 2013. A pathway approach to investigate the function and regulation of SREBPs. Genes Nutr 8, 289-300.
84. Jeon, T.I., Osborne, T.F., 2012. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab 23, 65-72.
85. Bennett, M.K., Osborne, T.F., 2000. Nutrient regulation of gene expression by the sterol regulatory element binding proteins: increased recruitment of gene-specific coregulatory factors and selective hyperacetylation of histone H3 in vivo. Proc Natl Acad Sci U S A 97, 6340-6344.
86. Lin, J., Yang, R., Tarr, P.T. y col. 2005. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 120, 261-273.
87. Li, S., Brown, M.S., Goldstein, J.L., 2010. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 107, 3441-3446.
88. Shimomura, I., Bashmakov, Y., Horton, J.D., 1999. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem 274, 30028-30032.
89. Düvel, K., Yecies, J.L., Menon, S. y col. 2010. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39, 171-183.
90. Yellaturu, C.R., Deng, X., Park, E.A. y col. 2009. Insulin enhances the biogenesis of nuclear sterol regulatory element-binding protein (SREBP)-1c by posttranscriptional down-regulation of Insig-2A and its dissociation from SREBP cleavage-activating protein (SCAP).SREBP-1c complex. J Biol Chem 284, 31726-31734.
91. Peterson, T.R., Sengupta, S.S., Harris, T.E. y col. 2011. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408-420.
92. Willy, P.J., Mangelsdorf, D.J., 1997. Unique requirements for retinoid-dependent transcriptional activation by the orphan receptor LXR. Genes Dev 11, 289-298.
93. Yoshikawa, T., Shimano, H., Amemiya-Kudo, M. y col. 2001. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol 21, 2991-3000.
94. Chen, G., Liang, G., Ou, J. y col. 2004. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci U S A 101, 11245-11250.
95. Ye, J., DeBose-Boyd, R.A., 2011. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol 3. pii: a0047
96. Bobard, A., Hainault, I., Ferré, P. y col. 2005. Differential regulation of sterol regulatory element-binding protein 1c transcriptional activity by insulin and liver X receptor during liver development. J Biol Chem 280, 199-206.
97. Horton, J.D., Bashmakov, Y., Shimomura, I., Shimano, H., 1998. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci U S A 95, 5987-599217.
98. Hasty, A.H., Shimano, H., Yahagi, N., Amemiya-Kudo, M. y col. 2000. Sterol regulatory element-binding protein-1 is regulated by glucose at the transcriptional level. J Biol Chem 275, 31069-31077.
99. Dentin, R., Pégorier, J.P., Benhamed, F. y col. 2004. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem 279, 20314-20326.
100. Liang, G., Yang, J., Horton, J.D. y col. 2002. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem 277, 9520-9528.
101. Nagai, Y., Yonemitsu, S., Erion, D.M. y col. 2009. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab 9, 252-264.
102. Lee, J.N., Zhang, X., Feramisco, J.D. y col. 2008. Unsaturated fatty acids inhibit proteasomal degradation of Insig-1 at a postubiquitination step. J Biol Chem 283, 33772-33783.
103. Kammoun, H.L., Chabanon, H., Hainault, I. y col. 2009. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest 119, 1201-1215.
104. Inoue, J., Ito, Y., Shimada, S. y col. 2011. Glutamine stimulates the gene expression and processing of sterol regulatory element binding proteins, thereby increasing the expression of their target genes. FEBS J 278, 2739-2750.
105. Xia, M., Liu, Y., Guo, H. y col. 2013. Retinol binding protein 4 stimulates hepatic sterol regulatory element-binding protein 1 and increases lipogenesis through the peroxisome proliferator-activated receptor-γ coactivator 1β-dependent pathway. Hepatology 58, 564-575.
106. Zhang, Y., Lei, T., Huang, J.F. y col. 2011. The link between fibroblast growth factor 21 and sterol regulatory element binding protein 1c during lipogenesis in hepatocytes. Mol Cell Endocrinol 342, 41-47.
107. Ruderman, N.B., Xu, X.J., Nelson, L. y col. 2010. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab 298, E751-760.
108. Bécard, D., Hainault, I., Azzout-Marniche, D. y col. 2001. Adenovirus-mediated overexpression of sterol regulatory element binding protein-1c mimics insulin effects on hepatic gene expression and glucose homeostasis in diabetic mice. Diabetes 50, 2425-2430.
109. Lemoine, M., Barbu, V., Girard, P.M. y col. 2006. Altered hepatic expression of SREBP-1 and PPARgamma is associated with liver injury in insulin-resistant lipodystrophic HIV-infected patients. AIDS 20, 387-395.
110. Eberlé, D., Clément, K., Meyre, D. y col. 2004. SREBF-1 gene polymorphisms are associated with obesity and type 2 diabetes in French obese and diabetic cohorts. Diabetes 53, 2153-2157.
111. Biddinger, S.B., Almind, K., Miyazaki, M. y col. 2005. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes 54, 1314-1323.
112. Teran-Garcia, M., Adamson, A.W., Yu, G. y col. 2007. Polyunsaturated fatty acid suppression of fatty acid synthase (FASN): evidence for dietary modulation of NF-Y binding to the Fasn promoter by SREBP-1c. Biochem J 402, 591-600.
113. Leavens, K.F., Easton, R.M., Shulman, G.I. y col. 2009. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab 10, 405-418.
114. Yecies, J.L., Zhang, H.H., Menon, S. y col. 2011. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 14, 21-32.
115. Moon, Y.A., Liang, G., Xie, X. y col. 2012. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab 15, 240-246.
116. Tang, J.J., Li, J.G., Qi, W. y col. 2011. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab 13, 44-56.
117. Xiao, X., Song, B.L., 2013. SREBP: a novel therapeutic target. Acta Biochim Biophys Sin (Shanghai) 45, 2-10.
118. Yamashita, H., Takenoshita, M., Sakurai, M., Bruick, R.K., Henzel, W.J., Shillinglaw, W., Arnot, D., Uyeda, K., 2001. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A 98, 9116-9121.
119. Foufelle, F., Gouhot, B., Pégorier, J.P. y col. 1992. Glucose stimulation of lipogenic enzyme gene expression in cultured white adipose tissue. A role for glucose 6-phosphate. J Biol Chem 267, 20543-20546.
120. Girard, J., Ferré, P., Foufelle, F., 1997. Mechanisms by which carbohydrates regulate expression of genes for glycolytic and lipogenic enzymes. Annu Rev Nutr 17, 325-352.
121. Ishii, S., Iizuka, K., Miller, B.C., Uyeda, K., 2004. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A 101, 15597-15602.
122. Iizuka, K., 2013. Recent progress on the role of ChREBP in glucose and lipid metabolism. Endocr J 60, 543-555.43.
123. Herman, M.A., Peroni, O.D., Villoria, J. y col. 2012. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484, 333-338.
124. Cairo, F., Rotundo, R., Frazzingaro, G. y col. 2001. [Diabetes mellitus as a risk factor for periodontitis]. Minerva Stomatol 50, 321-330.
125. Kawaguchi, T., Osatomi, K., Yamashita, H. y col. 2002. Mechanism for fatty acid "sparing" effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem 277, 3829-3835.
126. Ma, L., Robinson, L.N., Towle, H.C., 2006. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem 281, 28721-28730.55.
127. Kawaguchi, T., Takenoshita, M., Kabashima, T., Uyeda, K., 2001. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci U S A 98, 13710-13715.
128. Tsatsos, N.G., Towle, H.C., 2006. Glucose activation of ChREBP in hepatocytes occurs via a two-step mechanism. Biochem Biophys Res Commun 340, 449-456.
129. Bricambert, J., Miranda, J., Benhamed, F. y col. 2010. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest 120, 4316-4331.
130. Zeidan, Q., Hart, G.W., 2010. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci 123, 13-22.
131. Guinez, C., Filhoulaud, G., Rayah-Benhamed, F. y col. 2011. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60, 1399-1413.
132. Kabashima, T., Kawaguchi, T., Wadzinski, B.E., Uyeda, K., 2003. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci U S A 100, 5107-5112.57.
133. Dentin, R., Tomas-Cobos, L., Foufelle, F. y col. 2012. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J Hepatol 56, 199-209.
134. Arden, C., Tudhope, S.J., Petrie, J.L. y col. 2012. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem J 443, 111-123.
135. Cha, J.Y., Repa, J.J., 2007. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem 282, 743-751.
136. Gauthier, K., Billon, C., Bissler, M. y col. 2010. Thyroid hormone receptor beta (TRbeta) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. J Biol Chem 285, 28156-28163.
137. Iizuka, K., Bruick, R.K., Liang, G. y col. 2004. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A 101, 7281-7286.
138. Repa, J.J., Liang, G., Ou, J. y col. 2000. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14, 2819-2830.
139. Sirek, A.S., Liu, L., Naples, M. y col. 2009. Insulin stimulates the expression of carbohydrate response element binding protein (ChREBP) by attenuating the repressive effect of Pit-1, Oct-1/Oct-2, and Unc-86 homeodomain protein octamer transcription factor-1. Endocrinology 150, 3483-3492.
140. Havula, E., Hietakangas, V., 2012. Glucose sensing by ChREBP/MondoA-Mlx transcription factors. Semin Cell Dev Biol 23, 640-647.
141. Hurtado del Pozo, C., Vesperinas-García, G., Rubio, M. y col. 2011. ChREBP expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. Biochim Biophys Acta 1811, 1194-1200.
142. Benhamed, F., Denechaud, P.D., Lemoine, M. y col. 2012. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest 122, 2176-2194.
143. Dentin, R., Benhamed, F., Hainault, I. y col.2006. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 55, 2159-2170.
144. Iizuka, K., Takeda, J., Horikawa, Y., 2009. Hepatic overexpression of dominant negative Mlx improves metabolic profile in diabetes-prone C57BL/6J mice. Biochem Biophys Res Commun 379, 499-504.
145. Robichon, C., Girard, J., Postic, C., 2008. [Can the hyperactivity of lipogenesis cause hepatic steatosis? A role for ChREBP]. Med Sci (Paris) 24, 841-846.
146. Lodhi, I.J., Wei, X., Semenkovich, C.F., 2011. Lipoexpediency: de novo lipogenesis as a metabolic signal transmitter. Trends Endocrinol Metab 22, 1-8.
147. Charlton, M.R., 2013. ChREBP in NASH - a liver transcription factor comes in from the cold. J Hepatol 59, 178-179.
148. Färnegårdh, M., Bonn, T., Sun, S. y col. 2003. The three-dimensional structure of the liver X receptor beta reveals a flexible ligand-binding pocket that can accommodate fundamentally different ligands. J Biol Chem 278, 38821-38828.
149. Peet, D.J., Turley, S.D., Ma, W. y col. 1998. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 93, 693-704.
150. Blaschke, F., Takata, Y., Caglayan, E. y col. 2006. A nuclear receptor corepressor-dependent pathway mediates suppression of cytokine-induced C-reactive protein gene expression by liver X receptor. Circ Res 99, e88-99.
151. Willy, P.J., Umesono, K., Ong, E.S. y col. 1995. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev 9, 1033-1045
152. Zhang, Y., Repa, J.J., Gauthier, K., Mangelsdorf, D.J., 2001. Regulation of lipoprotein lipase by the oxysterol receptors, LXRalpha and LXRbeta. J Biol Chem 276, 43018-43024.
153. Korach-André, M., Parini, P., Larsson, L. y col. 2010. Separate and overlapping metabolic functions of LXRalpha and LXRbeta in C57Bl/6 female mice. Am J Physiol Endocrinol Metab 298, E167-178.
154. Korach-André, M., Archer, A., Barros, R.P. y col. 2011. Both liver-X receptor (LXR) isoforms control energy expenditure by regulating brown adipose tissue activity. Proc Natl Acad Sci U S A 108, 403-408.
155. Viennois, E., Pommier, A.J., Mouzat, K. y col. 2011. Targeting liver X receptors in human health: deadlock or promising trail? Expert Opin Ther Targets 15, 219-232.
156. Chen, J.D., Evans, R.M., 1995. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377, 454-457.
157. Glass, C.K., Rosenfeld, M.G., 2000. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14, 121-141.
158. Lee, S., Lee, J., Lee, S.K., Lee, J.W., 2008. Activating signal cointegrator-2 is an essential adaptor to recruit histone H3 lysine 4 methyltransferases MLL3 and MLL4 to the liver X receptors. Mol Endocrinol 22, 1312-1319.
159. Jakobsson, T., Venteclef, N., Toresson, G. y col. 2009. GPS2 is required for cholesterol efflux by triggering histone demethylation, LXR recruitment, and coregulator assembly at the ABCG1 locus. Mol Cell 34, 510-518.
160. Venteclef, N., Jakobsson, T., Ehrlund, A. y col. 2010. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev 24, 381-395.
161. Lehmann, J.M., Kliewer, S.A., Moore, L.B. y col. 1997. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem 272, 3137-3140.
162. Li, X., Yeh, V., Molteni, V., 2010. Liver X receptor modulators: a review of recently patented compounds (2007- 2009). Expert Opin Ther Pat 20, 535-562.
163. Ou, J., Tu, H., Shan, B. y col. 2001. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A 98, 6027-6032.
164. Oberkofler, H., Schraml, E., Krempler, F., Patsch, W., 2003. Potentiation of liver X receptor transcriptional activity by peroxisome-proliferator-activated receptor gamma co-activator 1 alpha. Biochem J 371, 89-96.
165. Li, X., Zhang, S., Blander, G. y col. 2007. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28, 91-106.
166. Yamamoto, T., Shimano, H., Inoue, N. y col.2007. Protein kinase A suppresses sterol regulatory element-binding protein-1C expression via phosphorylation of liver X receptor in the liver. J Biol Chem 282, 11687-11695.
167. Tobin, K.A., Ulven, S.M., Schuster, G.U. y col. 2002. Liver X receptors as insulin-mediating factors in fatty acid and cholesterol biosynthesis. J Biol Chem 277, 10691-10697.
168. Liu, X., Qiao, A., Ke, Y. y col. 2010. FoxO1 represses LXRα-mediated transcriptional activity of SREBP-1c promoter in HepG2 cells. FEBS Lett 584, 4330-4334.
169. Chinetti, G., Lestavel, S., Bocher, V. y col. 2001. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med 7, 53-58.
170. Brendel, C., Schoonjans, K., Botrugno, O.A. y col. 2002. The small heterodimer partner interacts with the liver X receptor alpha and represses its transcriptional activity. Mol Endocrinol 16, 2065-2076.
171. Kalaany, N.Y., Gauthier, K.C., Zavacki, A.M. y col. 2005. LXRs regulate the balance between fat storage and oxidation. Cell Metab 1, 231-244.
172. Repa, J.J., Turley, S.D., Lobaccaro, J.A. y col. 2000. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 289, 1524-1529.
173. Brunham, L.R., Kruit, J.K., Pape, T.D. y col. 2006. Tissue-specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises plasma HDL cholesterol levels. Circ Res 99, 672-674.
174. Feig, J.E., Pineda-Torra, I., Sanson, M. y col. 2010. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J Clin Invest 120, 4415-4424.
175. Cao, G., Liang, Y., Broderick, C.L. y col. 2003. Antidiabetic action of a liver x receptor agonist mediated by inhibition of hepatic gluconeogenesis. J Biol Chem 278, 1131-1136.
176. Grefhorst, A., van Dijk, T.H., Hammer, A. y col. 2005. Differential effects of pharmacological liver X receptor activation on hepatic and peripheral insulin sensitivity in lean and ob/ob mice. Am J Physiol Endocrinol Metab 289, E829-838.
177. Laffitte, B.A., Chao, L.C., Li, J. y col. 2003. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci U S A 100, 5419-5424.
178. Ross, S.E., Erickson, R.L., Gerin, I. y col. 2002. Microarray analyses during adipogenesis: understanding the effects of Wnt signaling on adipogenesis and the roles of liver X receptor alpha in adipocyte metabolism. Mol Cell Biol 22, 5989-5999.
179. Dalen, K.T., Ulven, S.M., Bamberg, K. y col. 2003. Expression of the insulin-responsive glucose transporter GLUT4 in adipocytes is dependent on liver X receptor alpha. J Biol Chem 278, 48283-48291.
180. Gerin, I., Dolinsky, V.W., Shackman, J.G. y col. 2005. LXRbeta is required for adipocyte growth, glucose homeostasis, and beta cell function. J Biol Chem 280, 23024-23031.
181. Pettersson, A.M., Stenson, B.M., Lorente-Cebrián, S. y col. 2013. LXR is a negative regulator of glucose uptake in human adipocytes. Diabetologia 56, 2044-2054.
182. Zitzer, H., Wente, W., Brenner, M.B. y col. 2006. Sterol regulatory element-binding protein 1 mediates liver X receptor-beta-induced increases in insulin secretion and insulin messenger ribonucleic acid levels. Endocrinology 147, 3898-3905.
183. Choe, S.S., Choi, A.H., Lee, J.W. y col. 2007. Chronic activation of liver X receptor induces beta-cell apoptosis through hyperactivation of lipogenesis: liver X receptor-mediated lipotoxicity in pancreatic beta-cells. Diabetes 56, 1534-1543.
184. López, J.M., Bennett, M.K., Sanchez, H.B. y col. 1996. Sterol regulation of acetyl coenzyme A carboxylase: a mechanism for coordinate control of cellular lipid. Proc Natl Acad Sci U S A 93, 1049-1053.
185. Tabor, D.E., Kim, J.B., Spiegelman, B.M., Edwards, P.A., 1998. Transcriptional activation of the stearoyl-CoA desaturase 2 gene by sterol regulatory element-binding protein/adipocyte determination and differentiation factor 1. J Biol Chem 273, 22052-22058.
186. Joseph, S.B., Laffitte, B.A., Patel, P.H. y col. 2002. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem 277, 11019-11025.
187. Chisholm, J.W., Hong, J., Mills, S.A., Lawn, R.M., 2003. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res 44, 2039-2048.
188. Grefhorst, A., Elzinga, B.M., Voshol, P.J. y col. 2002. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J Biol Chem 277, 34182-34190.
189. Ducheix, S., Montagner, A., Polizzi, A. y col. 2013. Essential fatty acids deficiency promotes lipogenic gene expression and hepatic steatosis through the liver X receptor. J Hepatol 58, 984-992.
190. Patel, R., Patel, M., Tsai, R. y col. 2011. LXRβ is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J Clin Invest 121, 431-441.
191. Ducheix, S., Montagner, A., Theodorou, V. y col. 2013. The liver X receptor: a master regulator of the gut-liver axis and a target for non alcoholic fatty liver disease. Biochem Pharmacol 86, 96-105.
192. Higuchi, N., Kato, M., Shundo, Y. y col. 2008. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol Res 38, 1122-1129.
193. Zelcer, N., Tontonoz, P., 2006. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest 116, 607-614.
194. Hong, C., Tontonoz, P., 2008. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr Opin Genet Dev 18, 461-467.
195. Jakobsson, T., Treuter, E., Gustafsson, J., Steffensen, K.R., 2012. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci 33, 394-404.
196. Wang, Y.Y., Dahle, M.K., Agren, J. y col. 2006. Activation of the liver X receptor protects against hepatic injury in endotoxemia by suppressing Kupffer cell activation. Shock 25, 141-146.
197. Mallat, A., Lotersztajn, S., 2011. The liver X receptor in hepatic stellate cells: a novel antifibrogenic target? J Hepatol 55, 1452-1454.
198. Beaven, S.W., Wroblewski, K., Wang, J. y col. 2011. Liver X receptor signaling is a determinant of stellate cell activation and susceptibility to fibrotic liver disease. Gastroenterology 140, 1052-1062.
199. Musso, G., Gambino, R., Cassader, M., 2013. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res 52, 175-191.
200. Caballero, F., Fernández, A., De Lacy, A.M. y col. 2009. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol 50, 789-796.
201. Steffensen, K.R., Jakobsson, T., Gustafsson, J., 2013. Targeting liver X receptors in inflammation. Expert Opin Ther Targets 17, 977-990.
202. Griffett, K., Solt, L.A., El-Gendy, B.l.-D. y col. 2013. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem Biol 8, 559-567.
203. Zhai, Y., Wada, T., Zhang, B., Khadem, S., Ren, S., Kuruba, R., Li, S., Xie, W., 2010. A functional cross-talk between liver X receptor-α and constitutive androstane receptor links lipogenesis and xenobiotic responses. Mol Pharmacol 78, 666-674.
204. Roth, A., Looser, R., Kaufmann, M. y col. 2008. Regulatory cross-talk between drug metabolism and lipid homeostasis: constitutive androstane receptor and pregnane X receptor increase Insig-1 expression. Mol Pharmacol 73, 1282-1289.
205. Eissing, L., Scherer, T., Tödter, K y col. 2013. De novo lipogenesis in human fat and liver is linked to ChREBP-ß and metabolic health. Nat Commun 4, 1528-1549.