ARTÍCULO |
Long-life supplementation with atenolol converts the heart and skeletal muscle unsaturation of mitochondrial membranes of mice into those of ten fold longer-lived mammals. Effects on mean and maximum longevity
Alexia Gómez1, A. Naudi2, R. Pamplona2 and Gustavo Barja1*
1Department of Animal Physiology II, Faculty of Biological Sciences, Complutense University, c/Antonio Novais n° 2, Madrid 28040, Spain.2Department of Experimental Medicine, University of Lleida-IRBLleida, E-25008 Lleida, Spain.
e-mail: gbarja@bio.ucm.es
Premio del Consejo General de Colegios Oficiales de Farmacéuticos del Concurso Científico de la Real Academia Nacional de Farmacia 2012. An. Real Acad. Farm. Vol 79, N° 2 (2013), pag. 253-273
abstract
The long-term effects of atenolol in drinking water throughout the whole lifespan (3.3 years) of a mammal (128 C57BL/6 male mice-SPF) were studied for the first time. We observed beneficial aging-related changes: decreases in the degree of unsaturation of mitochondrial membranes and of the 22:6n-3 fatty acid, an increase in oleic acid, as well as decreases in protein oxidation, glycoxidation and lipoxidation and oxidative damage in mtDNA in heart and skeletal muscle mitochondria. However, a secondary effect of the drug only in old animals was detected that agrees with recent meta-analyses in human patients. |
Keywords: Oxidative stress; β-blocker; Heart rate.
resumen
La suplementación de larga duración con atenolol convierte la insaturación de las membranas mitocondriales de músculo esquelético y de corazón de ratón en la característica de mamíferos de supervivencia diez veces mayor. Efectos sobre la longevidad media y máxima
Se estudia por primera vez el efecto a largo plazo del atenolol en el agua de bebida durante toda la vida (3,3 años) de un mamífero (128 ratones C57BL/6 macho-SPF). Observamos cambios beneficiosos relacionados con el envejecimiento: descenso en el grado de insaturación de las membranas mitocondriales y del ácido graso 22:6n-3, un incremento del ácido oleico, y descenso de la oxidación, glicoxidación y lipoxidación de proteínas y daño oxidativo al ADNmt en mitocondrias de corazón y músculo esquelético. Sin embargo, detectamos un efecto secundario del fármaco sólo en animales viejos que coincide con meta-análisis recientes en pacientes humanos. |
Palabras clave: Estrés oxidativo; β-bloqueante; Frecuencia cardíaca.
1. introduction
TA new mammalian longevity model based on ß-adrenergic receptor signaling interruption at the level of adenylyl cyclase has reported decreased bone and heart aging and increases in mean and maximum longevity in AC5 KO (adenylyl cyclase 5 Knockout) mice (1). We have previously mimicked this model with the ß-blocker atenolol in short-term studies (2), in which we have successfully modified one of the only two known traits correlating with longevity in the right sense: the low degree fatty acid unsaturation of the cellular membranes of the tissues of long-lived animals.
Comparative gerontological studies have already unveiled two traits that can explain the different (maximum) longevity of different mammals: long-lived animal species have a low rate of mitochondrial reactive oxygen species production (mitROSp) (3, 4) and a low unsaturation degree of membrane fatty acids (5, 6).
The first of these two factors, mitROSp, can be experimentally decreased with dietary manipulations like caloric restriction (7, 8), protein restriction (9) and methionine restriction (10). But the second one, the unsaturation degree of membrane fatty acids, is more difficult to modify. Increasing dietary saturated fatty acids have unhealthy effects on plasma cholesterol levels, and the tissue global FA (fatty acid) unsaturation is homeostatically regulated in mammalian tissues through control of gene expression (11). Deficiency of essential PUFAs in the diet leads to strong compensatory increases in tissue mead acid (20:3n-9, synthesized from 18:1n-9), a known diagnostic marker of essential FA deficiency, or to increases in MUFAs like 16:1n-7 and 18:1n-9 (12). Homeostatic changes like these are responsible for the failure to effectively change tissue DBI after feeding the animals with diets differing in FA unsaturation (13, 14).
A low unsaturation degree is most important for developing a high longevity because membrane fatty acid double bounds are most susceptible to oxidative attack due to two reasons: a) oxygen and many radical species are several times more soluble in lipid membrane bilayers than in the aqueous solution (15); b) the sensitivity of membranes to lipid peroxidation increases strongly as a function of the number of double bonds per fatty acid molecule. A lower total number of double bounds of membrane fatty acids make these molecules more resistant to lipid peroxidation. Polyunsaturated fatty acids (PUFAs) exhibit the highest sensitivity to ROS induced oxidative damage among cellular macromolecules, and this sensitivity increases as a function of the number of double bonds per fatty acid molecule (16,17). The negative relationship between membrane fatty acid composition and longevity has been observed in all the animal models studied, including mammals, birds, rodents, honeybees, mussels, and humans (18, 19): the longer the longevity of a species, the smaller the degree of unsaturation of the FAs present in their cellular membranes.
Recently, many mammalian models of extended lifespan caused single gene-mutations have been developed (20, 21). Most of them are related to insulin/IGF-1-like signaling pathways (22). But there are others like Agtr1a–/– (Angiotensine II type 1 receptors targeted disrupted) mice (23) and AC5KO (adenylyl cyclase 5 Knockout) mice (1) that also show increased medium and maximum longevity.
In the AC5KO model, extension of lifespan in 129/SvJ-C57BL/6 mice has been obtained through the disruption of β-adrenergic receptor signaling at the Type 5 adenylyl cyclase (AC5) level (1). This mouse showed increased mean and maximum longevity, from 25 to 33 months, and from 33 to 37 months, respectively, and also showed improvements in parameters related to bone and heart age-related deterioration. These improvements seem to be signaled to the nucleus through the Raf/ MEK/ extra cellular signal-regulated kinase (p-ERK) pathway, which was increased in heart and other tissues of the AC5KO mice, together with increases in the protein levels of MnSOD (manganese superoxide dismutase) in heart, kidney and brain, suggesting that a decrease in oxidative stress is involved in the mechanisms responsible for the aging delaying effect.
Two years ago we discovered that the AC5KO model can be mimicked, at least on the short-term, with the drug atenolol, by treating C57BL/6 normal mice during 15 days with this β1-selective blocker simply added to drinking water (2). In the present study we test the long-term effects of this drug given to mice in their drinking water throughout their whole life, using 128 male mice studied during their whole life span (more than 3 years) under SPF conditions. Based on the successful results of our previous short-term studies (2), we hypothesized that atenolol would chronically decrease the global degree of unsaturation of heart and skeletal muscle (SKM) mitochondrial membranes of mice to levels almost similar to those of species with one order of magnitude higher longevity, and would decrease specific markers of oxidative stress due to lowered in vivo lipid peroxidation. In this investigation it is tested for the first time whether modifying one of the only two known correlates of longevity (the double bond index of mitochondrial membranes, DBI) can decrease highly specific markers of oxidative stress in two post-mitotic tissues of a mammal and whether it can modify its longevity.
In those life-long atenolol-treated animals and their controls we studied physiological parameters including rectal temperature, basal metabolic rate, heart rate and blood pressure (at 18 and 35 months of age). Concerning oxidative stress, we measured the rate of ROS production in isolated functional mitochondrial (mtROSp), mitochondrial oxygen consumption in states 4 (resting) and 3 (phosphorylating), the amounts of the respiratory complex I, II, III and IV proteins, the apoptosis-inducing factor (AIF) which is also required for the assembly/maintenance of complex I (24), the marker of oxidative damage to mitochondrial DNA (mtDNA) 8-oxo-7,8-dihydro-2´-deoxyguanosine (8-oxodG), five different specific markers of protein oxidative modification: the specific protein carbonyls glutamic and aminoadipic semialdehydes (GSA and AASA) indicating purely protein oxidative modification, the protein glycoxidation markers carboxyethyl lysine (CEL) and carboxymethyl lysine (CML), and the protein lipoxidation markers CML and malondialdehyde lysine (MDAL). We also measured the full fatty acid composition of heart and SKM mitochondrial membranes which allowed us to calculate their global unsaturation indexes (the DBI, and the peroxidizability index, PI). The mitochondrial biogenesis indicators peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α (PGC1), the mitochondrial transcription factor (TFAM), antioxidant regulation transcription factor Nrf2 (Nuclear factor erythroid 2-related factor 2), and the p-ERK and SIRT1 signalling proteins related to longevity (25), as well as the basal metabolic rate of the whole animal, the rectal temperature, and the heart rate, the systolic and diastolic and mean arterial pressures were also measured.
2. MATERIALs and MeThODS
2.1. Animals and study design
128 C57BL/6 male mice were maintained in a separate room of Complutense University animal house at the Faculty of Medicine under SPF conditions during their whole life or until some of them were sacrificed to measure physiological or biochemical relevant parameters. 86 of these animals (43 Old control and 43 Old-AT-treated) were used only to obtain the survival curves. These animals were maintained under optimum conditions (12:12 (light-dark) cycle, 22°C ± 2°C and 50% ± 10% relative humidity), were left intact during their whole lifespan (except for the AT-treatment) and their day of spontaneous death was recorded. The treatment with atenolol was started at 2 months of age. A separate group of 42 animals (21 controls and 21 AT-treated) was established at the beginning of the experiment in order to measure the different physiological and biochemical parameters when reaching old age (after 16 months of experimentation). Among the 21 reporter animals of each of these two groups (control and AT-treated), 7 animals were used for the ROS and oxygen consumption measurements in isolated mitochondria, 7 animals were used for the measurement of 8-oxodG, and 7 animals were used to assay the rest of the biochemical parameters after being sacrificed by cervical dislocation. At the time of sacrifice these reporter animals had 18 months of age. The animals in the atenolol group had free access to a solution of 0.1 g/L of atenolol (Sigma, A7655) in drinking water. The diet (Panlab, Spain) was offered ad libitum to all animals. Just after cervical dislocation, hearts and SKM (total hind limb muscle) were immediately processed to isolate functional mitochondria, which were used to measure mitochondrial respiration and rates of mtROSp. Whole hearts and SKM tissue samples (total hind limb muscle) were stored at -80°C for the posterior analyses of the rest of the biochemical parameters. The experimental animal subject review committee from the Complutense University of Madrid approved all the experiments in C57BL/6 mice.
2.2. Physiological parameters
Rectal temperature was measured using a rectal probe (Thermocouple thermometer model 8112-20, Cole-Parmer Instrument Company). The measurements were performed three times in each mouse, always at 11:00, on three different days separated 15 days from each other, during the last 2 months of experimentation. To estimate the basal metabolic rate, individual mice we placed inside a closed-system respirometer (total volume 2,600 ml) and the carbon dioxide produced was captured with a 10% KCl solution. The rate of oxygen consumption of each animal was measured at rest with an oxygen analyzer and probe (Model 600 Can 1691, Engineered Systems & Designs) at 23±1°C. The measurements were realized at the end of the pharmacological treatment. Heart rate and blood pressure were measured in conscious mice with a noninvasive tail-cuff manometry system (LE5001 Panlab Harvard Apparatus). Each animal was acclimatized for at least three practice sessions in the three consecutive weeks before the final measurements were recorded. In each session 8 consecutive readings were recorded and their average was used to obtain systolic, diastolic, and mean blood pressure. These measurements were performed during the last 2 months of experimentation.
2.3. Isolation of functional mitochondria, oxygen consumption and ROS production
Mitochondria were obtained from fresh tissue by the procedure of Mela and Seitz (26) with modifications. After checking the functionality and phosphorylation capacity of the mitochondria (high respiratory control ratios) the rate of mtROSp was measured by the fluorometric method established at our laboratory (27). The rate of oxygen consumption of heart and SKM mitochondria was measured at 37°C in a water-thermostatized incubation chamber with a computer-controlled Clark-type O2 electrode (Oxygraph, Hansatech, UK) as previously described (28).
2.4. Oxidative damage to mtDNA (8-oxodG)
Isolation of mtDNA was performed by the method of Latorre and cols (29) adapted to mammals (30). The isolated mitochondrial DNA was digested to deoxynucleoside level and the level of oxidative damage in mtDNA was estimated by measuring the amount of 8-oxo-7,8-dihydro-2’deoxyguanosine (8-oxodG) referred to that of the non-oxidized base (deoxyguanosine, dG) by HPLC-EC as previously described (31).
2.5. Measurement of mitochondrial complexes I to IV, AIF, SIRT1, PGC1, TFAM, Nrf2 and ERK
The amounts of a) the mitochondrial respiratory chain complexes (I to IV), the complex I regulatory factor AIF, the mitochondrial biogenesis protein indicators SIRT1, PGC1, and TFAM, the Nrf2-ARE pathway marker Nrf2, and ERK1/2 pathway marker (ERK1/2 and phospho-ERK1/2) were estimated using western blot analyses as previously described (2).
2.6. Oxidation-derived protein damage markers
GSA, AASA, CML, CEL and MDAL were determined as trifluoroacetic acid methyl esters (TFAME) derivatives in acid hydrolyzed delipidated and reduced mitochondrial protein samples by GC/MS (32) using a HP6890 Series II gas chromatograph (Agilent, Barcelona, Spain) with a MSD5973A Series detector and a 7683 Series automatic injector, a HP-5MS column (30-m x 0.25-mm x 0.25-µm), and the described temperature program (32). The amounts of product were expressed as µmoles of GSA, AASA, CML, CEL or MDAL per mol of lysine.
2.7. Fatty acid analyses and global fatty acid unsaturation indexes
Fatty acids from mitochondrial lipids were analyzed as methyl esters derivatives by gas chromatography (GC) as previously described (33). The following fatty acyl indices were also calculated: saturated fatty acids (SFA); unsaturated fatty acids (UFA); monounsaturated fatty acids (MUFA); polyunsaturated fatty acids (PUFA) from n-3 and n-6 series (PUFAn-3 and PUFAn-6); and average chain length (ACL)=[(Σ%Total14 x 14) + (Σ% Total16×16) + (Σ%Total18×18) + (Σ%Total20×20) + (Σ% Total22×22) + (Σ% Total24×24)]/100. The density of double bonds in the membrane was calculated by the Double Bond Index, DBI = [(1×Σmol% monoenoic) + (2×Σmol% dienoic) + (3×Σmol% trienoic) + (4×Σmol% tetraenoic) + (5×Σmol% pentaenoic) + (6×Σmol% hexaenoic)]. Finally, the membrane susceptibility to peroxidation was calculated by the Peroxidizability Index, PI= [(0.025×Σmol% monoenoic) + (1×Σmol% dienoic) + (2×Σmol% trienoic) + (4×Σmol% tetraenoic) + (6×Σmol% pentaenoic) + (8×Σmol% hexaenoic)].
2.8. Statistics
Values were expressed as means ± standard error of the mean (SEM). Comparisons between groups were analyzed by ANOVA followed by DMS tests for paired groups and Student-t test. The survival curve was performed by Kaplan-Meier in Statgraphics, and mean and maximum (last 10% surviving individuals) life span values were analysed by the log rank test. The minimum level of statistical significance was set at P< 0.05 in all the analyses.
3. RESULTS
The mean body weight of the animals did not show significant differences between the two experimental groups at the beginning neither at the end of the experiment (results not shown).
No significant differences were observed between atenolol and control animals for rectal temperature (35.24±0.1 °C in Old Controls and 35.51±0.2 in Old AT) or basal metabolic rate (3.44±0.28 mlO2/g hr in Old Controls and 3.32±0.3 in Old AT).
Neither the basal rates of mtROSp of heart mitochondria (with glutamate/malate and with succinate+rotenone) nor the maximal ones (with glutamate/malate+rotenone) were significantly modified by atenolol treatment (results not shown). In the case of SKM mitochondria only a significant decrease with succinate+rotenone (from 0.92±0.07 in Old Controls to 0.75±0.05 in Old AT P<0.05) was detected. No significant differences in mitochondrial oxygen consumption were observed either, except for significant decreases only in the case of state 3 respiration in heart with both complex I –linked (glutamate/malate: 222.7±30.0 nanomoles of O2 /min mg mitochondrial protein in Old Control and 131.8±8.7 in Old AT, P<0.05) and complex II-linked (succinate+rotenone: 270.4±21.2 nanomoles of O2 /min mg mitochondrial protein in Old Control and 201.4±8.8 in Old AT) respiration.
Concerning respiratory complexes and AIF, no significant differences were observed for any parameter in the case of SKM (results not shown). In heart mitochondria, only the amount of complex II increased from 104.0±5.8 in Old Controls to 135.6±12.6 in Old AT (P<0.05; ratio of complex II/porin in arbitrary units), the rest of the parameters not showing significant variations (results not shown).
Atenolol treatment decreased the highly unsaturated 22:6n-3 FA and increased the much less unsaturated 18:1n-9 in heart and SKM (Figure 1). The decrease in 22:6n-3 was of 23% in heart and of 38% in SKM. Besides, the ratio 22:6n-3/24:6n-3, an index of the final steps of n-3 synthesis through β-peroxisomal lipoxidation (Figure 2) decreased in both tissues in the atenolol group (Figure 1; by 37% in SKM and by 46% in heart).
AT animals showed higher 18:1n-9 (large increase), 20:4n-6 and 24:5n-3, and lower 14:0, 20:3n-6 and especially 22:6n-3 than controls (18:1n-9 and 22:6n-3 values are shown in Figure 1 while the other FAs, which were measured, are not shown). In SKM, AT animals showed higher 18:1n-9 and 20:2n-6, and lower 22:6n-3 (large decrease) than controls. The increase in 18:1n-9 was of 30% in SKM and of 56% in heart. As a result of those changes, the global indexes of fatty acid unsaturation DBI and PI were strongly decreased by the atenolol treatment in both kinds of mitochondria (Figure 3). The DBI decreased by 22% in SKM and by 11% in heart mitochondria, while the PI was decreased by 31% in SKM and by 17% in heart mitochondria.
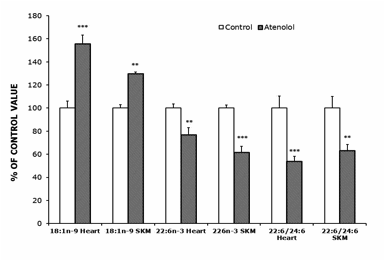
Figure 1.- Oleic (18:1n-9), Docosahexanoic (22:6n-3) and Peroxisomal Beta-Oxidation (22:6/24:6 ratio) from control and atenolol treated mice. Values are means ± SEM from 5-6 (heart) or 6 (SKM) different animals and are expressed as percentage of those in the controls for each parameter. Control values: 12.85±0.76 (18:1n-9, heart); 28.03±1.01 (22:6n-3, heart); 187.94±19.46 (22:6/24.6, heart); 16.39±0.47 (18:1n-9, SKM); 21.29±0.53 (22:6n-3, SKM); 103.44±10.32 (22:6/24.6, SKM). Asterisks represent significant differences between the control and the atenolol group. * P<0.05; ** P<0.01; *** P<0.001.
 Figure 2.- Metabolic pathway
from fatty acids of the n-3 serie. The name of the mouse enzymes likely to
contribute to elongation (ELOVL 2 and 5) and desaturation (Stearoyl-CoA
desaturase, SCDs) steps are indicated in these pathway.
Figure 2.- Metabolic pathway
from fatty acids of the n-3 serie. The name of the mouse enzymes likely to
contribute to elongation (ELOVL 2 and 5) and desaturation (Stearoyl-CoA
desaturase, SCDs) steps are indicated in these pathway.
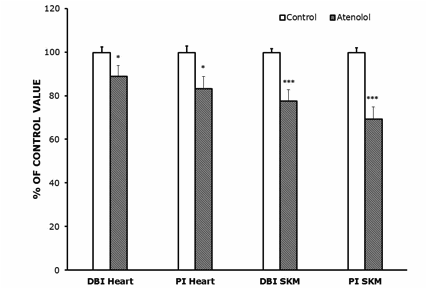
Figure 3.- Double bond index (DBI) and peroxidizability index (PI) in heart (A) and SKM (B) mitochondrial fatty acids from control and atenolol treated mice. Values are means ± SEM from 6 (heart) or 5-6 (SKM) different animals and are expressed as percentage of those in the controls for each parameter. Control values: 232.60±5.90 (DBI, heart); 268.39±8.02 (PI, heart); 203.91±3.38 (DBI, SKM); 217.36±4.52 (PI, SKM). For calculation of DBI and PI values see the Materials and Methods section. Asterisks represent significant differences between the control and the atenolol group. * P<0.05; ** P<0.01; *** P<0.001.
Oxidative damage in heart mtDNA significantly decreased from 20.65±3.81 8-oxodG/105dG in Old Controls to 10.07±1.37 in Old AT (P<0.05), whereas in the case of SKM the trend to decrease in the AT group did not reach statistical significance (results not shown).
Protein oxidation, glycoxidation and lipoxidation markers are shown in Figures 4 and 5. In heart mitochondria all the five markers measured, GSA, AASA, CEL, CML and MDAL, were significantly lower in Old AT-animals than in Old controls (Figure 4). In SKM mitochondria the values of GSA, AASA, CML and MDAL were significantly lower in the atenolol treated animals. These decreases were rather strong and ranged from 31% to 51% depending on the parameter measured and the tissue considered. Only in the case of CEL the decrease shown by AT compared to controls did not reach statistical significance (Figure 5). Concerning mitochondrial biogenesis, antioxidant factors and signaling proteins, SIRT1 increased and Nrf2 decreased in Old AT in heart (P<0.05) but not in SKM mitochondria. TFAM decreased in Old AT in SKM (P<0.05) but not in heart mitochondria, while PGC1 did not show significant changes in any organ (results not shown). The ratio of the phosphorylated to total ERK (p-ERK/total ERK) showed significantly higher values in the atenolol than in the control group (Figure 6).
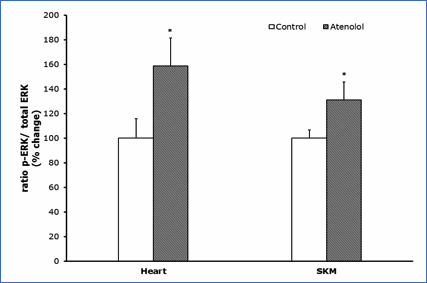
Figure 4.- Protein oxidation, glycoxidation and lipoxidation indicators in heart mitochondria from control and atenolol treated mice. Values are means ± SEM from 6 different animals and are expressed as percentage of those in the controls for each protein modification marker. Control values: 3,817±128 (glutamic semialdehyde, GSA); 434±37 (AASA, aminoadipic semialdehyde, AASA); 557±41 (carboxyethyl-lysine, CEL); 1038±77 (carboxymethyl-lysine, CML); 16295±364 (malondialdehyde-lysine, MDAL). Units: µmol/mol lysine. Asterisks represent significant differences between the control and the atenolol group. ** P<0.01; *** P<0.001.
No significant differences in heart rate, or the systolic, mean and diastolic arterial blood pressures were observed at 18 months of age (Figure 7A). In contrast, the heart rate measured at 35 months of age was significantly and strongly decreased in the atenolol group (Figure 7B), whereas the systolic, mean and diastolic arterial pressures trends to decreased values did not reach statistical significance (Figure 7B).
Finally, the Kaplan Meier´s survival curve (Figure 8) showed a similar mean life span (50% survival) and a lower maximum (at 10% survival) longevity in the atenolol group only at old age, the difference in survival starting only after 28 months of age.
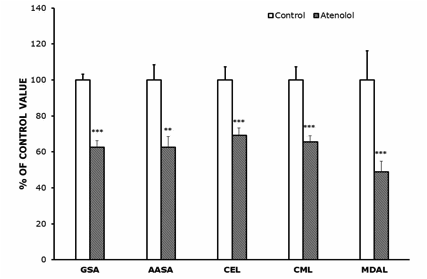
Figure 5.- Protein oxidation, glycoxidation and lipoxidation indicators in SKM mitochondria from control and atenolol treated mice. Values are means ± SEM from 6 different animals and are expressed as percentage of those in the controls for each protein modification marker. Control values: 4,415±272 (glutamic semialdehyde, GSA); 463±24 (AASA, aminoadipic semialdehyde, AASA); 380±25 (carboxyethyl-lysine, CEL); 770±48 (carboxymethyl-lysine, CML); 632±65 (malondialdehyde-lysine, MDAL). Units: µmol/mol lysine. Asterisks represent significant differences between the control and the atenolol group. ** P<0.01; *** P<0.001.
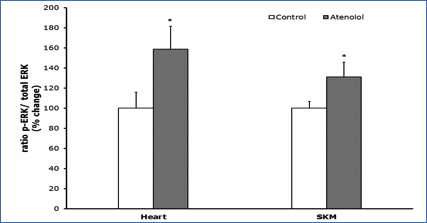 Figure 6.- Ratio p-ERK/total ERK. Ratio of phosphorylated
versus total ERK (nonphosphorylated) (p-ERK/ERK); ERK represents the sum of the
44-kD and the 42-kD ERK proteins. Values presented are means_standard error of
the mean (SEM) from n=4 different animals per group over immunoreactivity in
control values. Asterisk represents significant differences compared to values
of untreated animals. (*) p<0.05.
Figure 6.- Ratio p-ERK/total ERK. Ratio of phosphorylated
versus total ERK (nonphosphorylated) (p-ERK/ERK); ERK represents the sum of the
44-kD and the 42-kD ERK proteins. Values presented are means_standard error of
the mean (SEM) from n=4 different animals per group over immunoreactivity in
control values. Asterisk represents significant differences compared to values
of untreated animals. (*) p<0.05.
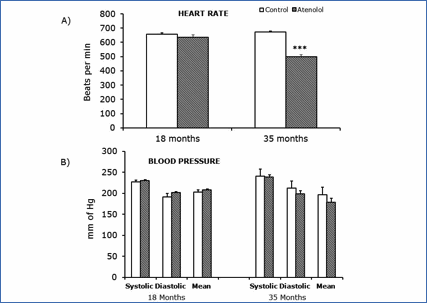
Figure 7.- Blood pressure parameters (B) and heart rate (A) measured at 18 and 35 months. Values are means ± SEM from 7 different animals. Blood pressure units: mm of Hg Heart rate units: bits per min. (***) p<0.001.
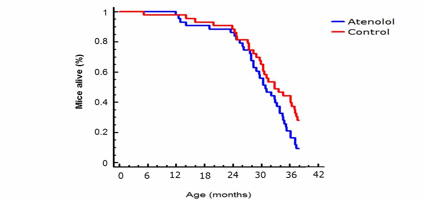
Figure 8.- Kaplan-Meier survival curve for control and atenolol-treated C57BL6 male mice. (log-rank χ2= 4.40401, P= 0.0358512) life span extension in control mice (n= 43 for control and n= 43 for atenolol mice).
4. discusSion
In the present work we comprehensively studied, for the first time, the chronic effect of the β-1 blocker atenolol on various metabolic and oxidative stress parameters during the whole life span of a sufficiently large population of healthy mice.
After 16 months of continuous atenolol treatment, we observed quantitatively important decreases in the fatty acid unsaturation degree of mitochondrial membranes, as well as in protein oxidation, lipoxidation and glycoxidation in mitochondria from the two tissues studied: heart and SKM. This agrees with similar changes observed after 15 days of treatment with the same dose of atenolol in mouse heart (2), indicating that the most important capacity of this drug to lower this parameter can be maintained at least during most of the lifespan of the animals.
These results indicate that blocking of the β1-adrenergic signaling pathway improves one of the only two known parameters which link longevity and oxidative stress, the DBI and PI (reviewed in 34, 35). Strikingly, the potency of atenolol is so great that it can decrease the DBI of mitochondrial membranes from that of a mouse to that typical of a mammal of around 40 years in longevity (2), taking into account the known relationship between membrane FA unsaturation and mammalian longevity (34), also in relation to the extended life-span of the AC5KO mice (1).
In our longevity experiment, the atenolol treatment did not modify body weight, heart and SKM organ weight, or food intake, this ruling out the possibility that the observed changes could be secondary effects of caloric restriction. In our study, atenolol treatment did not change either complex I or III mtROS generation rate neither with glutamate/malate nor with pyruvate/malate as substrates or with the supplemented complex I and III specific inhibitors rotenone or antimicyn A. These results agree with those from our short-term study in the heart of atenolol-treated C57BL/6 mice (2), and are in contrast to dietary, protein and methionine restriction models in which mtROSp decreases at complex I (36). All of these dietary manipulations increase longevity and decrease mtROSp without changing the DBI and PI (37). However, the β-adrenergic signalling blockade seems to decrease the other longevity-related trait (lowers the DBI and PI) without changing mtROSp. Therefore, these two manipulations, the dietary restrictions and the atenolol treatment, seem to be complementary.
Supporting that general idea, long-term treatment with atenolol did not change any of the mitochondrial respiratory chain complexes except for the increase in complex II (70 KDa subunit, Flavoprotein) in heart mitochondria, mtROSp and the level of 8-oxodG in mtDNA (which indicates the balance between mtDNA oxidative damage and repair). mtROSp and 8-oxodG in mtDNA usually change together and in similar direction in different models of dietary restriction studied and both are lower in long-lived compared to short-lived animal species (38). In the present study, although the atenolol treatment did not decrease mtROSp, the oxidative damage to mtDNA was significantly lower in heart mitochondria in the atenolol group and tended to be lower (non significant trend) in SKM mitochondria. Rarely, but sometimes, the changes in both parameters (mtROSp and mtDNA oxidative damage) in longevity modifying experiments have been apparently dissociated (39). Maybe the longest time of drug exposure of the present study can induce an increase in the mitochondrial mtDNA repair systems, resulting in the decreased 8-oxodG levels observed.
There is a systematic negative relationship between tissue membrane fatty acid unsaturation and longevity in all mammals studied to date (35, 40). Extraordinarily long-lived animals like birds (41, 42), naked mole rats (43), the echidna (44) and queen honey bees (35) also show a common trait: they also have a low fatty acid unsaturation degree in their tissue cellular membranes. This makes their membranes more resistant to lipid peroxidation, since the sensitivity of membrane lipids to lipid peroxidation increases in an exponential way as a function of the number of double bonds per fatty acid molecule (17). This also occurs in long-lived wild-derived strains of mice when compared to genetically heterogeneous laboratory mice (45). In our study, the atenolol treatment significantly decreased the DBI in heart and SKM (11% and 22.35% respectively total decrease) and the PI also in both tissues (16.76% and 30.66% respectively total decrease). These results are in general agreement with our previous study in C57BL/6 mice heart, in which the decrease was 40% for the PI and 30% for the DBI respectively, although this was observed, at variance with the present investigation, in total heart tissue instead of in heart and SKM mitochondria (2).
The longevity-related decrease in global FA unsaturation is due to a redistribution between the type of PUFAs present from the highly unsaturated docosahexaenoic (22:6n-3) and sometimes arachidonic (20:4n-6) acids in short-lived animal species to the less unsaturated linoleic acid (18:2n-6) and, in some cases, linolenic acid (18:3n-3) in the long-lived ones at mitochondrial and tissue levels (40). Among these FAs, the one contributing most to the low global fatty acid unsaturation of long-lived animals is 22:6n-3. This agrees strikingly well with our results, which show an important decrease in the amount of docosahexahenoic acid (22:6n-3) in the atenolol treated animals in mitochondria from both tissues. It is also interesting that the fatty acid showing quantitatively more important increases in long lived mammals in general is 18:2n-6, but in the bird case, important increases occurs frequently for the monounsaturated oleic acid (18:1n-9) in long-lived species (46), a FA with well known beneficial effects in many nutritional studies, and this FA also increases in our case in the long-life atenolol-treated group.
Docosahexahenoic acid (22:6n-3) has six double bonds and consequently has five bis-allylic hydrogens per chain, and is 320-times more susceptible to ROS attack than oleic acid (18:1n-9), which is consistent with the strong decrease in secondary protein lipoperoxidation observed (lower MDAL and CML in AT-treated animals). In our case, a most relevant factor that contributed to decrease the DBI and PI seems to be the strong decrease in β-peroxisomal lipoxidation (estimated as the 22:6n-3/24:6n-3 ratio) in the atenolol group. The main function of this process seems to be the partial degradation of very-long chain fatty acids, producing chain-shortened acyl-CoAs, acetyl-CoA and NADH, which may exit from peroxisomes via pores that permit the influx of substrates and efflux of products of β-oxidation. These substrates go back to the mitochondria to complete the fatty acid oxidation process (47).
The decrease in DBI and PI confers higher resistance of membranes to lipid peroxidation and lowers lipoxidation-dependent damage to macromolecules, like proteins, and (likely) mtDNA. The long-term atenolol treatment was able to very strongly and significantly decrease protein oxidation (GSA and AASA), glycoxidation (CEL and CML) and lipoxidation (CML and MDAL) markers in both tissues, except for CEL in SKM which also showed a trend to decrease that did not reach statistical significance. Aging is known to increase protein oxidation in association with a functional decline of proteasome activity (48) whereas decreases in protein oxidation and increases in the catabolism of modified proteins have been described in experimental modifications that extend longevity, like dietary restriction (49) and methionine restriction (50, 33) even when applied to old animals (51). The decreased fatty acid unsaturation degree most likely leads to a lower lipid-derived secondary free radical formation, decreased specific protein oxidation and damage to other macromolecules (52) which was reflected, in our case, in the decrease in protein oxidation, glycoxidation and lipoxidation, as well as, in the case of the heart, mtDNA oxidative damage.
The molecular mechanism suggested to explain these changes could be the following: binding of hormones and neurotransmitters to β-adrenergic receptors activates adenylate cyclase (AC) increasing cyclic adenosine monophosphate (cAMP) and then protein kinase A (PKA). PKA inhibits Raf-1, which, in turn, stimulates p-MEK and p-ERK. p-ERK enters the nucleus, where it can modify gene expression through the action of many different molecules. Because AC stimulates PKA, and PKA inhibits Raf-1, an increase in the Raf/MEK/ERK pathway is expected when AC is lacking or β-adrenergic receptors are blocked. In agreement with this, an increase in p-MEK and p-ERK was observed in tissues of AC5KO mice, including the heart (1). The same happens in our pharmacological model of β-adrenergic blockade by atenolol, in which p-ERK levels were increased both at short-term in the mice heart (2), as well as in the present study after long-life AT treatment in heart and SKM mitochondria. This protein can enter the nucleus and activate different transcription factors, modifying genes related to oxidative stress, including genes coding for desaturase and elongase enzymes, as well as those controlling peroxisomal β-oxidation, and are rate limiting for the synthesis of the highly peroxidizable 22:6n-3 FA. Lower desaturase/elongase/peroxisomal β-oxidation activities induced by the AT-blockade (via low AC and high p-ERK) would decrease 22:6-n3 formation from its less unsaturated 18:3n-3 dietary precursor.
Concerning cellular signaling, AMPK responds to high intracellular levels of AMP, and activates (besides others) the expression of SIRT1 (Silent information regulator 1) (53). SIRT1 regulates energy metabolism, cell apoptosis, cell proliferation and inflammation, as well as stress resistance by means of FOXO, p53 and NF-B signaling, increasing the intracellular concentration of NAD+. SIRT1 is increased in caloric restriction (life-extending) models, and can activate cellular stress resistance, playing an anti-aging role (54). In our study, SIRT1 levels were higher after the atenolol treatment in the heart, which indicates that the blocking of AC inactivates the AMPc. That would increase SIRT1 expression through the ensuing changes in intracellular levels of AMP then of AMPK.
Nrf2 is the “master regulator” of the antioxidant response modulating the expression of many several antioxidant-codifying genes (55), and TFAM is a regulator of mtDNA transcription, whose lack leads to severe respiratory chain deficiency (56). Since it is now well known that long-lived animals have lower tissue levels of antioxidant enzymes and other endogenous antioxidants (57) and less endogenous DNA base excision repair (BER) activity (58), which are secondary events to the lower rate of mtROSp of long-lived animal species (59, 37), it is not strange that Nrf2 and TFAM were decreased after atenolol treatment.
Finally, although our results show an improvement in parameters related with longevity, a low DBI, PI, protein oxidation and lipoxidation in mitochondria from both tissues, and in mtDNA oxidative damage (in the case of heart), this was not enough to increase longevity, as it is evident form the survival curves finally obtained, since atenolol treated mice did not live longer than the control animals. Although mean life span was similar in both groups, only at the end of the life span and in very old animals (equivalent to 70-80 years old humans) survival was somewhat decreased after long-term treatment with atenolol. This can be due to a deleterious secondary effect of the drug. All β-blockers act by decreasing blood pressure/heart rate (60), and that is known to be advantageous for coronary disease patients, or for those surviving after heart attacks or other serious cardiovascular illnesses. However, recent meta-analyses in humans are suggesting that in the case of old hypertensive patient’s atenolol can decrease instead of increase survival (61). When old patients are treated with β-blockers (atenolol is used in around 75% of cases) rigid arteries typical of old people can result in sporadically too low diastolic or systolic blood pressures, which, together with the aged myocardium of old people can increase sudden cardiac death and thus mortality. It seems that the most important secondary effect of atenolol is the strongly decrease in heart rate that it induces (62, 63). The meta analyses recently performed in humans agree with our results in mice, because we did not see any differences in blood pressure or heart rate when measured at 18 months of age, whereas heart rate was strongly and significantly decreased in the atenolol group at 35 months of age (499 mmHg in Old AT compared to 674 mmHg in Old controls). We believe that our results are most important concerning future cardiologic treatments in humans since there are available antihypertensive drugs alternative to atenolol, and they constitute the first study of the long-life effects of atenolol in a large population of healthy mammal.
5. Conclusion
1.- The long-life treatment with the β-blocker atenolol in drinking water of a large population of mice strongly decreases the degree of fatty acid unsaturation of skeletal muscle and mitochondrial membranes, the only parameter known, apart from a low rate of mitochondrial ROS generation, that correlates with longevity in the right way.
2.- That change seems to be due to a large extent to a decrease in desaturase/elongase/peroxisomal β-oxidation activities, which are rate limiting for the synthesis of the highly unsaturated docosahexahenoic acid (22:6n-3). This fatty acid is universally avoided in the tissue cellular membranes of long-lived mammalian and bird species.
3.- Those decreases are due to β-adrenergic blockade-induced increases in p-ERK signaling to the nucleus, and lead to strong decreases in oxidation, glycoxidation and lipoperoxidation of mitochondrial proteins, as well as in oxidative damage to mtDNA (in the case of heart tissue).
4.- These beneficial changes were not translated into increased (maximum) longevity most likely due to a secondary detrimental effect of the drug on heart frequency, and likely eventual falls in arterial pressure, only evident in very old animals, whereas the mean life span was not altered. These effects are most relevant for consideration of possible detrimental effects of atenolol, recently detected (meta-analyses from 2008 on) only in old hypertensive human subjects.
6. acknowledgments
This investigation was supported in part by I+D grants from the Spanish Ministry of Science and Innovation (BFU2008-00335/BFI) to G.Barja.
7. REFERENCeS
1. Yan, L.; D.E. Vatner, D.E.; O’Connor, J.P.; Ivessa, A.; Ge, H.; Chen, W.; Hirotani, S.; Ishikawa, Y.; Sadoshima, J.; Vatner, S.F. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell, 2007; 130, 247–258.
2. Sanchez-Roman, I.; Gomez, J.; Naudi, A.; Ayala, V.; Portero-Otín, M.; Lopez-Torres, M.; Pamplona, R.; Barja, G. The beta-blocker atenolol lowers the longevity-related degree of fatty acid unsaturation, decreases protein oxidative damage, and increases extracellular signal-regulated kinase signaling in the heart of C57BL/6 mice. Rejuv Res 2010; 13, 683–693
3. Barja, G.; Cadenas, S.; Rojas, C.; Pérez-Campo, R.; López-Torres, M. Low mitochondrial free radical production per unit O2 consumption can explain the simultaneous presence of high longevity and high aerobic metabolic rate in birds. Free Radic Res 1994, 21, 317–327
4. Barja, G.; Mitochondrial oxygen consumption and reactive oxygen species production are independently modulated: implications for aging studies. Rejuv Res 2007; 10, 215–224
5. Pamplona, R.; Portero Otín, M.; Riba, D.; Ruiz, C.; Prat, J.; Bellmunt, M.J.; Barja, G. Mitochondrial membrane peroxidizability index is inversely related to maximum life span in mammals. J Lipid Res 1998; 39, 1989-94
6. Hulbert, A.J.; Pamplona, R.; Buffestein, R.; Buttemer, W.A. Life and death: metabolic rate, membrane composition and life span of animals. Physiological Reviews 2007; 87, 1175-1213
7. Hagopian, K.; Chen, Y.; Simmons Domer, K.; Soo Hoo, R.; Bentley, T.; McDonald, R.B.; Ramsey, J.J. Caloric restriction influences hydrogen peroxide generation in mitochondrial sub-populations from mouse liver. J Bioenerg Biomembr 2011; 43, 227-36
8. Gredilla, R.; Barja, G. Caloric restriction, aging and oxidative stress. Endocrinology 2005; 146, 3713–3717
9. Sanz, A.; Caro, P.; Barja, G. Protein restriction without strong caloric restriction decreases mitochondrial oxygen radical production and oxidative DNA damage in rat liver. J Bioenerg Biomembr 2004; 36, 545–552
10. Sanz, A.; Caro, P.; Ayala, V.; Portero-Otin, M.; Pamplona, R.; Barja, G. Methionine restriction decreases mitochondrial oxygen radical generation and leak as well as oxidative damage to mitochondrial DNA and proteins. FASEB J 2006a; 20, 1064–1073
11. Maresca, B., Cossins, A.R. Fatty acid feedback and fluidity. Nature 1993; 365, 606–607.
12. Hoch, F.L. Cardiolipins and membrane function. Biochim. Biophys Acta 1992; 1113, 71–133.
13. Pamplona, R.; Portero-Otín, M.; Sanz, A.; Requena, J.; Barja, G. Modification of the longevity-related degree of fatty acid unsaturation modulates oxidative damage to proteins and mitochondrial DNA in liver and brain. Experimental Gerontology 2004; 39, 725–733
14. Sato, A., Huang, M.Z., Watanabe, S., Okuyama, H., Nakamoto, H., Rada´k, Z., Goto, S. Protein carbonyl content roughly reflects the unsaturation of lipids in skeletal muscle but not in other tissues of stroke-prone spontaneously hypertensive strain (SHRSP) rats fed different fats and oils. Biol. Pharm. Bull. 1998; 21, 1271–1276.
15. Moreau, R.; Nguyen, BT.; Doneanu, CE.; Hagen, T.M. Reversal by aminoguanidine of the age-related increase in glycoxidation and lipoxidation in the cardiovascular system of Fischer 344 rats. Biochem Pharmacol, 2005; 69, 29–40.
16. Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free Radical Oxidation of Polyunsaturated Lipids: New Mechanistic Insights and the Development of Peroxyl Radical Clocks. Acc Chem Res, 2011; 44, 458-67.
17. Holman, R.T. Autoxidation of fats and related substances. In: Holman RT, Lundberg WO, Malkin T (eds) Progress in chemistry of fats and other lipids. Pergamon Press, London, 1954; 51–98
18. Hulbert, A.J. Metabolism and longevity: Is there a role for membrane fatty acids? Integr Comp Biol 2010; 50, 808-17
19. Naudi, A.; Jove, M.; Ayala, V.; Portero-Otin, M.; Barja, G.; Pamplona, R. Regulation of membrane unsaturation as antioxidant adaptive mechanism in long-lived animal species. Free Rad Antiox 2011; 1, 3-12.
20. Liang, H.; Masoro, Nelson, J.F.; Strong, R.; McMahan, C.A.; Richardson, A. Genetic mouse models of extended lifespan. Exp Gerontol, 2003; 38, 1353–1364
21. Selman, C.; Withers, D.J. Mammalian models of extended healthy lifespan. Philos Trans R Soc Lond B Biol Sci 2011; 366, 99-107
22. Narasimhan, S.D.; Yen, K.; Tissenbaum, H.A. Converging pathways in lifespan regulation. Curr Biol, 2009; 19, 657-6
23. Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; Morigi, M.; Coffman, T.M.; Remuzzi, G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 2009; 119, 524–530
24. Porter, A.G.; Urbano, A.G. Does apoptosis-inducing factor (AIF) have both life and death functions in cells? Bioessays 2006; 28, 834-843
25. Donmez, G.; Guarente, L. Aging and disease: connections to sirtuins. Aging Cell 2010; 9, 285-290
26. Mela, L.; Seitz, S. Isolation of mitochondria with emphasis on heart mitochondria from small amounts of tissue. Methods Enzymol 1997; 55, 39-46
27. Barja, G.; The quantitative measurement of H2O2 generation in isolated mitochondria. J Bioenerg Biomembr 2002; 34, 227-233
28. Gredilla, R.; Sanz, A.; Lopez-Torres, M.; Barja, G. Caloric restriction decreases mitochondrial free radical generation at Complex I and lowers oxidative damage to mitochondrial DNA in the rat heart. FASEB J 2001; 15, 1589-1591
29. Latorre, A.; Moya, A.; Ayala, A. Evolution of mitochondrial DNA in Drosophila suboscura. Proc Natl Acad Sci USA 1986; 83, 8649-8653
30. Asuncion, J.C.; Millan, A.; Pla, R.; Bruseghini, L.; Esteras, A., Pallardo, F.V.; Sastre, J.; Viña, J. Mitochondrial glutathione oxidation correlates with age-associated oxidative damage to mitochondrial DNA. FASEB J 1996; 10, 333-338
31. Barja G.; Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals, FASEB J, 2000; 14, 312-318
32. Pamplona, R.; Dalfó, E.; Ayala, V.; Bellmunt, MJ.; Prat, J.; Ferrer, I.; Portero-Otín, M. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J Biol Chem. 2005; 280(22), 21522-30.
33. Caro, P.; Gomez, J.; Sanchez, I.; Naudi, A.; Ayala, V.; López-Torres, M.; Pamplona, R.; Barja, G. Forty percent methionine restriction decreases mitochondrial oxygen radical production and leak at complex I during forward electron flow and lowers oxidative damage to proteins and mitochondrial DNA in rat kidney and brain mitochondria. Rejuvenation Res. 2009; 12(6), 421-34.
34. Pamplona, R.; Barja, G.; Portero-Otín, M. Membrane fatty acid unsaturation, protection against oxidative stress, and maximum life span: a homeoviscous-longevity adaptation. Ann New York Acad Sci 2002; 959, 475-490
35. Hulbert, A.J.; Pamplona, R.; Buffestein, R.; Buttemer, W.A. Life and death: metabolic rate, membrane composition and life span of animals. Physiological Reviews 2007; 87, 1175-1213
36. López-Torres, M.; G. Barja, G. Lowered methionine ingestion as responsible for the decrease in rodent mitochondrial oxidative stress in protein and dietary restriction. Possible implications for humans. Biochim Biophys Acta 2008; 1780, 1337-1347
37. Pamplona, R.; Barja, G. An evolutionary comparative scan for longevity-related oxidative stress resistance mechanisms in homeotherms. Biogerontology 2011; 12, 409:35
38. Pamplona, R.; Barja, G. Highly resistant macromolecular components and low rate of generation of endogenous damage: two key traits of longevity. Ageing Res Rev 2007; 6, 189–210
39. Caro P., Gómez J., Sanz A., Portero-Otín M., Pamplona R., Barja G. Effect of graded corticosterone treatment on aging-related markers of oxidative stress in rat liver mitochondria. Biogerontology 2007; 8, 1–11
40. Pamplona, R.; Barja, G.; Portero-Otín, M. Membrane fatty acid unsaturation, protection against oxidative stress, and maximum life span: a homeoviscous-longevity adaptation. Ann New York Acad Sci 2002; 959, 475-490
41. Pamplona, R.; Portero-Otín, M.; Requena, J.R.; Thorpe, S.R.; Herrero, A.; Barja, G. A low degree of fatty acid unsaturation leads to lower lipid peroxidation and lipoxidation-derived protein modification in heart mitochondria of the longevous pigeon than in the short-lived rat. Mech Ageing Dev 1999; 106, 283-296.
42. Buttemer, W.A.; Battam, H.; Hulbert, A.J. Fowl play and the price of petrel: long-living Procellariformes have peroxidation-resistant membrane composition compared with short-living Galliformes. Biol Lett 2008; 4, 351-354
43. Mitchell, T.W.; Buffenstein, R.; Hulbert, A.J.; Membrane phospholipid composition may contribute to exceptional longevity of the naked mole-rat (Heterocephalus glaber): A comparative study using shotgun lipidomics. Exp Gerontol 2007; 42, 1053-1062.
44. Hulbert, A.J.; Beard, L.A.; Grigg, G.C. The exceptional longevity of an egg-laying mammal, the short-beaked echidna (Tachyglossus aculeatus) is associated with peroxidation-resistant membrane composition. Exp Gerontol 2008; 43, 729-733
45. Hulbert, A.J.; Faulks, S.C.; Harper, J.M.; Miller, R.A.; Buffenstein, R. Extended longevity of wild-derived mice is associated with peroxidation-resistant membranes. Mech Ageing Dev 2006; 127, 653-657
46. Pamplona, R.; Prat, J.; Cadenas, S.; Rojas, C.,Pérez-Campo, R.; L6pez-Torres, M.; Barja G. Low fatty acid unsaturation protects against lipid peroxidation in liver mitochondria from long-lived species: the pigeon and human case. Mechanisms of Ageing and Development 1996; 53, 86 53-66
47. Vance, D.E.; Vance, J. Biochemistry of Lipids, Lipoproteins and Membranes; Bernardi, G., Ed.; New Comprehensive Biochemistry; ELSEVIER, 1996; 31, p 91
48. Kastle, M.; Grune, T. Protein oxidative modification in the aging organism and the role of the ubiquitin proteasomal system. Curr Pharm Des 2011; 17, 4007–4022
49. Dhabi, J.M.; Monte, P.L.; Wingo, J.; Rowley, B.C.; Cao, S.X.; Waldford, R.L.; Spindler, S.R. Caloric restriction alters the feeding response of key metabolic enzyme genes. Mech Ageing Dev 2001; 122, 1033–1048
50. Caro, P.; Gómez, J.; López-Torres, M.; Sánchez, I.; Naudí, A.; Jove, M.; Pamplona, R.; Barja, G. Forty percent and eighty percent methionine restriction decrease mitochondrial ROS generation and oxidative stress in rat liver. Biogerontology 2008; 9, 183–196
51. Sanchez-Roman, I.; Gómez, A.; Pérez, I.; Sanchez, C.; Suarez, H.; Naudí, A.; Jové, M.; Lopez-Torres, M.; Pamplona, R.; Barja G. Effects of aging and methionine restriction applied at old age on ROS generation and oxidative damage in rat liver mitochondria, Biogerontol. 2012; 13, 399-411.
52. Spiteller, G. Is lipid peroxidation of polyunsaturated fatty acids the only source of free radicals that induce aging and age-related diseases? Rejuv Res 2010; 13, 91-103
53. Salminena, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Research Reviews 2012¸11, 230– 241
54. Le Bourg, E. Hormesis, aging and longevity. Biochim. Biophys. Acta. 2009; 1790, 1030–1039.
55. Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord; J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Molecular Aspects of Medicine 2011; 32, 234–246
56. Dogan, S.A.; Trifunovic, A. Modelling Mitochondrial Dysfunction in Mice. Physiol. Res. 2011; 60 (Suppl. 1), S61-S70
57. Perez-Campo, R.; López-Torres, M.; Cadenas, S.; Rojas, C.; Barja, G. The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. J Comp Physiol B 1998; 168, 149–158
58. Page, M.M.; Stuart J.A. Activities of DNA base excision repair enzymes in liver and brain correlate with body mass, but not with lifespan, Age, 2011; 34, 1195-209.
59. Barja, G. Free radicals and aging. Trends in Neurosci 2004; 27, 595-600
60. Cockcroft, J.R.; Pedersen, M.D. b-Blockade: Benefits Beyond Blood Pressure Reduction? The Journal of Clinical Hypertension. 2012; 14, 112-120
61. Bangalore, S.; Sawhney, S.; Messerli, F.H. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J. Amer. College of Cardiol. 2008; 52, 1482-1489.
62. Cucherat, M.; Borer, J.S. Reduction of Resting Heart Rate With Antianginal Drugs: Review and Meta-Analysis. American Journal of Therapeutics 2012; 19, 269–280
63. London, G.M.; Asmar, R.G.; O’Rourke, M.F.; Safar M.E. Mechanism(s) of Selective Systolic Blood Pressure Reduction After a Low-Dose Combination of Perindopril/Indapamide in Hypertensive Subjects: Comparison With Atenolol. Journal of the American College of Cardiology. 2004, 43, 92-99.