SESIONES
CIENTÍFICAS |
Sesión científica conmemorativa del Premio Nobel 2012 de Fisiología o Medicina y del Premio Nobel de Química
 Juan
Ramón Lacadena Calero
Juan
Ramón Lacadena Calero
Coordinador de la sesión.
Sesión celebrada el 13 de diciembre de 2012
e-mail: edicion@ranf.com
ORDEN DEL DÍA
Presentación:
Excmo. Sr. D. Juan-Ramón Lacadena Calero
Académico de Número de la Real Academia Nacional de Farmacia
Ponentes:
“Una familia de receptores de membrana esencial para la comunicación celular”
Prof. Dr. Federico Mayor Menéndez
Catedrático de Inmunología, Facultad de Medicina, UAM. Director Científico, Instituto de Investigación Sanitaria Princesa. Hospital Universitario de la Princesa
“Rebobinando la película genética del desarrollo"
Excmo. Sr. D. Juan-Ramón Lacadena Calero
Premios Nobel de Química 2012: Una familia de receptores de membrana esencial para la comunicación celular
Federico Mayor Menéndez
Departamento de Biología Molecular y Centro de Biología Molecular “Severo Ochoa” (CSIC-Universidad Autónoma de Madrid), Universidad Autónoma, 28049 Madrid (España).
e-mail: fmayor@cbm.uam.es
Recibido el 18 de febrero de 2013 An. Real Acad. Farm. Vol 79, Nº 1 (2013), pag. 132-150.
RESUMEN
El premio Nobel de Química 2012 ha sido otorgado a los investigadores estadounidenses Robert J. Lefkowitz y Brian K Kobilka por sus estudios sobre los receptores acoplados a proteínas G. Esta familia de proteínas de membrana son los sensores biológicos más extendidos y versátiles, responsables en buena medida de la capacidad de nuestras células de recibir mensajes del entorno. Son también la diana de numerosos fármacos utilizados para el tratamiento de múltiples patologías. El trabajo de Lefkowitz ha sido decisivo para desvelar la naturaleza química de estos receptores y para entender mejor sus mecanismos de señalización y de regulación. Lefkowitz y Kobilka consiguieron también identificar el gen que codificaba para el receptor beta-adrenérgico, lo que permitió posteriormente desvelar la existencia de múltiples receptores de características similares. Finalmente, Kobilka ha utilizado la difracción por rayos X para determinar la estructura íntima de estas proteínas. El camino abierto por Lefkowitz y Kobilka permitirá conocer mejor las alteraciones de receptores en situaciones patológicas y avanzar en el diseño de nuevas estrategias terapéuticas. |
Palabras clave: Receptores; Proteínas G; Señalización celular.
ABSTRACT
A essential family for cellular communication membrane receptors
The Nobel Prize in Chemistry 2012 has been awarded to Robert J. Lefkowitz and Brian K. Kobilka for their studies on G-protein-coupled receptors. The components of this family of membrane proteins are ubiquitous and versatile biological sensors that play an essential physiological role by allowing our cells to respond to external stimuli. GPCR also are very important pharmacological targets for the treatment of a variety of pathological conditions. The contributions of Lefkowitz have been decisive in unveiling the chemical nature of these receptors and to better understand their signaling and regulatory mechanisms. Lefkowitz and Kobilka also cloned the beta-adrenergic receptor gene and paved the way for the identification of a large family of structurally-related receptor proteins. Finally, Kobilka has recently determined the tridimensional structure of prototypical GPCRs. The work by Lefkowitz and Kobilka opens exciting avenues for a better knowledge of receptor alterations in pathological situations and for the design of novel therapeutic strategies. |
Keywords: Receptors, G proteins, Cell signaling.
1. Introducción
El premio Nobel de Química 2012 ha reconocido los decisivos estudios de Robert Lefkowitz y Brian Kobilka en la identificación y caracterización de los denominados "receptores acoplados a proteínas G" (conocidos en el ámbito científico como GPCR, por las siglas correspondientes a “G protein-coupled receptors” en idioma inglés), de amplísima relevancia fisiológica y farmacológica (1). En este caso concreto, debo decir que a la satisfacción de todo científico cuando se reconocen los méritos de unos colegas, se suma un componente más personal, ya que conozco muy directamente a los dos premiados, desde que fui discípulo del Profesor Lefkowitz durante mi estancia en Duke University (Carolina del Norte) en los años 1985-1986, periodo en el que Brian Kobilka también formaba parte de su laboratorio. La superfamilia de receptores acoplados a proteínas G constituye un elemento central en las redes de señalización celular, por las que se transfiere información biológica del entorno, y cuyo conocimiento es esencial para entender el funcionamiento de los seres vivos y en particular de los organismos multicelulares.
2. Comunicación celular: capacidad de respuesta a cambios en el entorno
Todas las células deben recibir continuamente información del ambiente que las rodea, y tomar decisiones basadas en esa información. Así, los organismos unicelulares necesitan distinguir los nutrientes que se encuentran en su cercanía, y regular sus procesos metabólicos de acuerdo con esas disponibilidades.
En el caso de las células de los organismos multicelulares, es preciso que integren la información procedente de las células vecinas y del conjunto del organismo, para tomar decisiones tales como reproducirse, especializarse, moverse a otro sitio o morir. Por tanto, comprender cómo las células reciben y coordinan señales del entorno y de otras células del mismo organismo es esencial para entender procesos biológicos básicos (como la proliferación, diferenciación y apoptosis), la organización en tejidos, el metabolismo, la migración de las células o la propia percepción sensorial.
Antes de la aparición de los organismos multicelulares, los organismos unicelulares ya habían desarrollado mecanismos para responder a cambios en el medio y a detectar en él la presencia de otras células. El advenimiento de organismos multicelulares durante el proceso evolutivo precisó del desarrollo de nuevos sistemas de control de la actividad celular, del establecimiento de normas estrictas que regulasen el funcionamiento de cada una de las células especializadas del organismo para el beneficio del conjunto.
Además, debía asegurarse que la puesta en marcha de respuestas celulares se coordinase de tal manera que todas las células implicadas en un proceso biológico reaccionasen al unísono durante el desarrollo embrionario o ante respuestas fisiológicas. La solución evolutiva a estas necesidades de “socialización” celular fue el desarrollo de un “lenguaje” muy elaborado de comunicación, capaz no sólo de captar las señales externas (particularmente a través de los sistemas de percepción sensorial como la vista o el olfato), sino de integrar la información procedente de las células vecinas y del conjunto del organismo, mediante el establecimiento de rutas complejas de señalización que coordinasen y ejecutasen las respuestas celulares ante cambios ambientales, metabólicos o patogénicos del organismo.
A nivel molecular, los sistemas de señalización celular y de control de la expresión génica controlan el flujo de información desde el DNA a RNA, y de éste a proteínas (en los procesos de transcripción y traducción, respectivamente) y , en niveles de integración superior, regulan dinámicamente el interactoma (las múltiples redes de interacciones que establecen las proteínas y que sustentan las funciones celulares) y la función fisiológica integrada en el organismo global, resultado de la coordinación de todas esas funciones celulares , lo que se ha llamado “fisioloma”. La extraordinaria tarea de estos procesos de coordinación de la actividad celular resulta evidente si se considera que un ser humano adulto consta de aproximadamente 80-100 millones de millones de células, de unos 300 tipos celulares distintos, agrupadas en distintos tejidos y órganos, formando entre sí una intricada red de conexiones funcionales.
Mensajeros, receptores y cascadas de señalización intracelular: los GPCR como la familia de receptores más extendida y versátil
Los sistemas de señalización son extraordinariamente complejos, asemejándose a complicadas redes o circuitos con múltiples elementos de intersección y control. En general, estos sistemas se basan en la existencia de moléculas (denominadas mensajeros, hormonas, neurotransmisores, o mediadores químicos locales según su origen celular, forma de liberación, y función) que llevan “órdenes” sólo a aquellas células que poseen receptores específicos para reconocer a esa molécula.
Los receptores tienen la capacidad de actuar como detectores de señales y de transformar ese acto de reconocimiento molecular en una señal intracelular (denominada “segundo mensajero”). Los segundos mensajeros, ya desde dentro de la célula, modifican la actividad, localización o interacciones entre proteínas celulares (controlando así el metabolismo, o la función del citoesqueleto, por ejemplo) y también regulan la expresión génica, promoviendo una respuesta celular específica e integrada (Figura 1). Por tanto, los sistemas de señalización celular están normalmente organizados en etapas secuenciales de detección, transformación, amplificación y diseminación de la señal, que son un poderoso instrumento para el control de las principales funciones celulares.
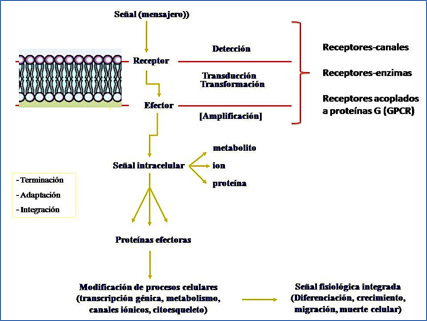
Figura 1.- Organización de las cascadas de señalización controladas por receptores situados en la membrana plasmática.
Es importante recordar que estos sistemas, para ser eficaces, tienen que funcionar de forma transitoria y controlada de tal forma que sólo persista la señal mientras lo haga el mensajero. Por tanto, tienen que existir además procesos de terminación, adaptación e integración que aseguren en todo momento su activación y desactivación controlada.
La mayoría de los mensajeros se unen a receptores situados en la membrana plasmática de las células diana. Se distinguen tres grandes clases de receptores, definidos por su “estrategia” para transformar la señal extracelular en una intracelular (Figura 1):
· Receptores acoplados a canales iónicos. Son proteínas de membrana que dejan pasar iones a través de ellas sólo si está presente el mensajero en la parte extracelular. Estos receptores actúan como “compuertas” que se abren transitoriamente dejando pasar calcio, sodio o cloruro (dependiendo del receptor) a favor de su gradiente electroquímico. Este tipo de receptores (constituidos por varias subunidades de proteínas transmembrana) son especialmente abundantes en células excitables caracterizadas por una gran rapidez de respuesta, como las neuronas o las células musculares.
· Receptores con actividad enzimática propia. Son en general proteínas con una región transmembrana que presentan un sitio de unión del mensajero situado en el exterior de la célula y una zona con actividad enzimática (sitio catalítico) en el interior. La unión del mensajero provoca cambios en el receptor (que en muchos casos implica su dimerización, es decir, su cercanía a otro receptor similar) que resultan en la estimulación de su actividad catalítica. Muy frecuentemente esa actividad es de tipo quinasa, es decir, provoca la fosforilación en un residuo de tirosina, de serina o de treonina de la propia proteína receptora y de otras proteínas celulares, modificando transitoriamente su función. Muchos mensajeros de tipo peptídico, como la insulina, y muchos factores que controlan el crecimiento de las células utilizan este tipo de receptores.
· Receptores acoplados a proteínas G. En las dos tipos de receptores anteriores, la capacidad de reconocer al mensajero y de promover y amplificar una señal intracelular (a través de un canal iónico o una actividad enzimática) residían en la misma proteína. En este último tipo participan 3 proteínas de membrana distintas: el receptor (que es en este caso una proteína que atraviesa 7 veces la membrana), unas proteínas transductoras denominadas proteínas G (situadas en la periferia interna de la membrana plasmática) y otra proteína efectora o amplificadora. Entre estas proteínas efectoras se encuentran la adenilil ciclasa (que produce el segundo mensajero denominado AMP cíclico a partir del ATP celular), fosfolipasas (que “liberan” segundos mensajeros “almacenados” en forma de lípidos en la membrana), o canales para diversos iones. Este sistema de señalización ha tenido un gran “éxito evolutivo”, ya que es la familia de receptores más extensa, más ubicua y más versátil. Los humanos tenemos casi mil receptores de esta familia en nuestro genoma (1-4), capaces de reconocer específicamente a un repertorio extremadamente variado de estímulos, desde fotones en la retina a múltiples aromas en el epitelio olfativo, pasando por receptores para aminas biógenas, aminoácidos, derivados lipídicos, péptidos y proteínas en múltiples tejidos, que controlan múltiples aspectos de la función celular y de la homeostasis del organismo (Figura 2).
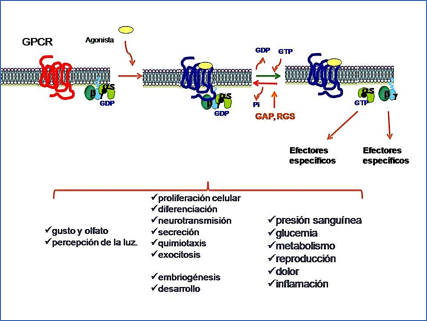
Figura 2.- Señalización mediada por receptores acoplados a proteínas G. La llegada del agonista promueve la interacción del receptor con la proteína G y el intercambio de GDP por GTP, lo que a su vez conduce a la disociación de las subunidades de la proteína G heterotrimérica, que pueden entonces interaccionar con efectores específicos y controlar una gran variedad de procesos celulares básicos, la percepción sensorial y diversos aspectos de la homeostasis del organismo. La activación de las proteínas G es transitoria, ya que su capacidad GTPasa (estimuladas por las proteínas GAP o RGS) vuelve el sistema a su situación basal.
Las proteínas G son interruptores moleculares que se activan transitoriamente. Estas proteínas pueden encontrarse en dos conformaciones espaciales diferentes: una forma inactiva, cuando unen al nucleótido GDP, y otra forma activada capaz de unirse con otras proteínas celulares denominadas efectoras, cuando unen GTP. Pero esta activación es intrínsecamente transitoria, ya que estas proteínas son GTPasas, es decir, destruyen al cabo de un breve tiempo el GTP transformándolo de nuevo en GDP, y vuelven así a su estado basal. Tanto el encendido (intercambio de GDP por GTP) como el apagado (hidrólisis de GTP) de este interruptor molecular se pude modular por su interacción con otras proteínas.
Hoy sabemos que cuando los GPCR reconocen a su mensajero (por ejemplo, el receptor de adrenalina a la adrenalina), cambian su conformación y pueden entonces interaccionar con una proteína G de tipo heterotrimérico unida a GDP, lo que a su vez promueve el intercambio de GTP por GDP. La proteína G en su estado activo interacciona con efectores (como la adenilil ciclasa) modificando parámetros intracelulares que diseminan la señal extracelular. Las subunidades Gbetagamma que se liberan simultáneamente pueden también actuar sobre diversos efectores celulares (Figura 2) Posteriormente, la proteína G hidroliza GTP a GDP (en un proceso que puede ser activado por familias de proteínas estimuladoras de la actividad GTPasa, denominadas GAP o RGS (5,6) y el sistema vuelve a su conformación basal. Sólo si sigue habiendo mensajero en el exterior de la célula se repetirá el ciclo de activación y desactivación.
Los GPCR son también muy relevantes por sus implicaciones fisiopatológicas y en farmacología (1,4,7,8). En muchas enfermedades se encuentran alterados los niveles de mensajeros y/o las rutas de señalización que controlan GPCRs. Por ejemplo, en patologías cardiovasculares existen aumentos en los niveles de mensajeros como catecolaminas, angiotensina o endotelina, que alteran a su vez el normal funcionamiento y crecimiento de tipos celulares cardiovasculares, y pueden conducir a hipertrofia cardiaca y a fallo cardiaco. La gran capacidad de control de las funciones celulares de los GPCR puede aprovecharse para modificarla de la forma más eficaz y específica posible.
Así, pueden seleccionarse o diseñarse compuestos químicos capaces de unirse con gran afinidad a los mismos receptores que nuestros mensajeros internos, consiguiendo así mimetizar (agonistas) o impedir (antagonistas) su acción. Por ejemplo, agonistas de receptores beta2-adrenérgicos son eficaces broncodilatadores y se utilizan para tratar el asma; antagonistas beta1-adrenérgicos se utilizan para el tratamiento de la hipertensión; antagonistas del receptor H2 de la histamina inhiben la excesiva secreción gástrica; agonistas de receptores de opiáceos, como la morfina, se utilizan como analgésicos, etc.
3. Evolución del concepto de receptor
El concepto de receptores como elementos sensores del entorno se remonta a Paul Ehrlich en el año 1903, cuando se avanzó la idea de que las sustancias biológicamente activas podrían unirse a sitios específicos en las superficies de las células. Posteriormente, en la primera década del siglo XX, JN Langley y su estudiante Henry Dale fueron los primeros en proponer explícitamente la idea de una sustancia receptora en las células capaces de responder a estímulos, basados en experimentos clásicos de fisiología y farmacología, utilizando preparaciones de músculo esquelético o liso y de glándulas salivales para estudiar los efectos de la adrenalina o la acetil-colina (9, 10). Sin embargo, la naturaleza físico-química de estos receptores era desconocida. En la década de 1940 el farmacólogo Raymond Ahlquist, examinando las diferentes reacciones de órganos a la adrenalina y sustancias químicas relacionadas, introdujo el concepto de la existencia de subtipos de receptores adrenérgicos (11): unos cuyo efecto principal es la contracción de células de músculo liso vascular (receptores alfa-adrenérgicos) y otros que estimulan la contracción cardiaca (beta-adrenérgicos).
Posteriormente, científicos como James Black (Premio Nobel de Fisiología o Medicina en el año 1988) desarrollaron sustancias capaces de interferir con la acción de la adrenalina y la noradrenalina en el corazón, los denominados fármacos beta-bloqueantes, que tuvieron una extraordinaria repercusión en el tratamiento de la enfermedad coronaria y la hipertensión.
A pesar de estos importantes progresos, a principios de los años 1960 existía todavía un conocimiento muy escaso de las características moleculares de los receptores y de los mecanismos de transmisión de la señal. En esta década se produjo un avance conceptual crítico, la teoría del segundo mensajero (12) sugerida por Earl Sutherland (que obtuvo el Premio Nobel en el año 1971). Sutherland había trabajado con el nobel Carl Cori estudiando los mecanismos por los que la adrenalina regula la degradación de glucógeno a glucosa en el hígado. Más adelante descubrió que para promover este efecto la adrenalina no entra en la célula sino que estimulaba la síntesis en el otro lado de la membrana de AMP cíclico (AMPc), tras la activación de una enzima adenilil ciclasa.
El AMPc actúa entonces como “segundo mensajero” transmitiendo la señal a proteínas intracelulares mediante la activación de una proteína quinasa dependiente de este nucleótido cíclico, identificada por Edwin Krebs (13).
En el marco de la teoría de la regulación alostérica que habían propuesto en el año 1963 Monod, Changeux y Jacob, una hipótesis muy atractiva era que la adenilil-ciclasa fuese una enzima alostérica de membrana con dos sitios diferentes, uno receptor en el exterior y otro catalítico en el interior de la célula. Sin embargo, los científicos estadunidenses Martin Rodbell y Alfred Gilman demostraron en la década de los 70 y principios de los 80 del siglo pasado que la realidad era más compleja, y que la actividad adenilil-ciclasa requería la presencia e hidrólisis de GTP, y que existían unas proteínas transductoras, denominadas proteínas G, que actuaban como intermediarios entre el reconocimiento de la adrenalina por su receptor y la estimulación de la actividad adenilil-ciclasa (14, 15). Estos científicos compartieron el Premio Nobel de Fisiología o Medicina en el año 1994.
En este contexto es en el que el trabajo de Robert Lefkowitz dio un impulso decisivo al entendimiento de la naturaleza y mecanismo de acción de los receptores de adrenalina. Lefkowitz, nacido en el año 1943 en el barrio de Bronx en Nueva York, se había formado como cardiólogo en la Universidad de Columbia y había realizado, tras un periodo de actividad clínica, una estancia postdoctoral en los Institutos Nacionales de la Salud con J. Roth e I. Pastan, trabajando en la identificación de las acciones de la hormona ACTH utilizando ensayos de radioligandos.
Sin embargo, cuando se estableció en la Universidad de Duke como investigador independiente en el año 1973, centró sus esfuerzos en los receptores beta-adrenérgicos, quizá por su formación como cardiólogo y unido al hecho de que su padre había fallecido recientemente a causa de una patología cardiaca. Durante los siguientes años, el laboratorio de Lefkowitz desarrolló una seria de técnicas que fueron esenciales para demostrar la existencia de los receptores como entidades moleculares diferenciadas, su cuantificación, y posterior purificación (Figura 3).
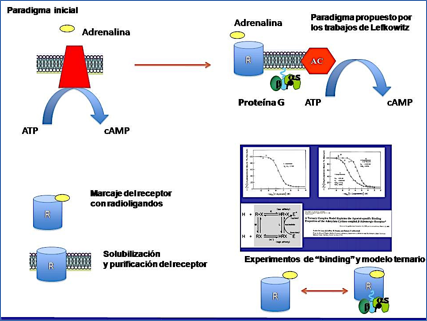
Figura 3.- Contribuciones esenciales del laboratorio de Lefkowitz en la identificación del receptor de adrenalina y de los mecanismos de activación de la adenilil-ciclasa.
La utilización de ligandos del receptor beta-adrenérgico marcados radioactivamente permitió la detección y cuantificación del receptor en las membranas celulares (16). Posteriormente, los ensayos de unión de ligando en diferentes condiciones experimentales permitieron identificar interacciones alostéricas complejas entre los receptores y las proteínas G. Así, la afinidad de los ligandos por el receptor beta-adrenérgico podía modularse por la presencia de GTP, mientras que la presencia de un agonista aumentaba la interacción entre el receptor y la proteína G. Estos experimentos llevaron a Lefkowitz, De Lean, Limbird y Stadel a proponer el denominado "modelo del complejo ternario", en el que el receptor activado unido a la proteína G constituía el elemento estimulador de la actividad adenilil-ciclasa (17-19).
Otro paso esencial en la caracterización del receptor beta-adrenérgico fue su solubilización y purificación. Con alguna excepción, como es la caso de la rodopsina en la retina, los receptores de membrana se encuentran generalmente en muy pequeñas concentraciones, lo que dificulta mucho su aislamiento y purificación. Gracias a la utilización de detergentes como la digitonina, Marc Caron y Lefkowitz consiguieron solubilizar un receptor funcional en el año 1976 (20).
A continuación Marc Caron, desarrolló métodos de cromatografía de afinidad basados en el antagonista beta-adrenérgico alprenolol, que permitieron la purificación de los receptores purificados en columnas de alprenolol-sefarosa a principios de los años 80 (21,22). La disponibilidad del receptor purificado permitió hacer un experimento conceptualmente crítico (23): Lefkowitz, en colaboración con Eva Neer y Lutz Birnbaumer, realizó experimentos de reconstitución de receptores purificados, proteínas G purificadas y la actividad catalítica de la adenilil-ciclasa, demostrando definitivamente que el mecanismo de transmisión de la señal de adrenalina incluía tres entidades moleculares diferentes.
Clonaje del receptor beta-adrenérgico y el nacimiento de una familia de receptores de membrana
El propio Bob Lefkowitz ha recordado en entrevistas realizadas en los últimos años que 1986 marcó un punto de inflexión crítico en su investigación (24). En efecto, entonces tuvo lugar el paso decisivo de identificar el gen que codificaba para el receptor beta-adrenérgico, lo que permitió también conocer la secuencia y características de los aproximadamente 400 aminoácidos que componen esa proteína.
Ese proyecto lo lideraba en su laboratorio un postdoctoral de extraordinaria perseverancia y talento, llamado Brian Kobilka. Este investigador, nacido en Little Falls (Minnesota) en 1955 y formado también como médico cardiólogo en la Universidad de Yale, se había incorporado como postdoctoral en la Universidad de Duke en el año 1984. La disponibilidad de receptor beta-2-adrenérgico purificado permitió intentar el clonaje del gen y cDNA de este receptor utilizando técnicas de microsecuenciación peptídica, para diseñar luego oligonucleótidos degenerados para intentar identificar el cDNA del receptor en genotecas de DNA genómico. Después de varios años de esfuerzo el grupo de Kobilka y Lefkowitz (en colaboración con Cathy Strader en Merck) consiguió publicar la secuencia completa del receptor beta-2 adrenérgico de hámster en el número de la revista Nature de mayo de 1986 (25). Sorprendentemente, el receptor de la adrenalina presentaba notables similitudes con el receptor de la luz (la rodopsina), en el sentido de que ambos parecían presentar siete tramos de aminoácidos capaces de atravesar la membrana celular (Figura 4).
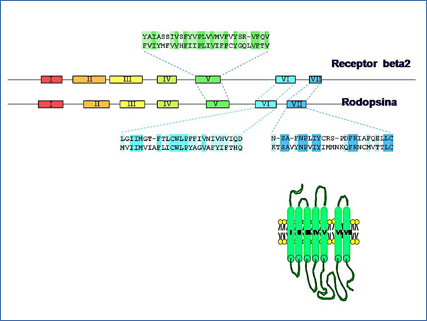
Figura 4.- Clonaje del receptor beta adrenérgico y concepto de familia de receptores acoplados a proteínas G. El conocimiento de la secuencia del receptor beta-2-adrenérgico permitió su comparación con la de la rodopsina e identificar similitudes en su estructura global y en la secuencia de sus dominios transmembrana, lo que permitió proponer la existencia de una familia de receptores de 7 dominios transmembrana con similares rasgos estructurales.
Al mismo tiempo, el laboratorio de Lefkowitz también descubrió que los mecanismos de regulación del receptor de adrenalina eran muy parecidos a los de la rodopsina de la retina. Se había descrito que en presencia de luz la rodopsina activada se fosforilaba en su dominio intracelular por una enzima denominada rodopsina quinasa, lo que promovía su desensibilización. También en 1986, Benovic, Mayor, Caron y Lefkowitz publicaron un artículo en Nature (26) en el que identificaban que una quinasa similar, denominada quinasa del receptor beta adrenérgico (βARK por sus siglas en inglés) era capaz de fosforilar al receptor beta-2 adrenérgico en respuesta a adrenalina, y también a la rodopsina en respuesta a la presencia de luz (Figura 5).
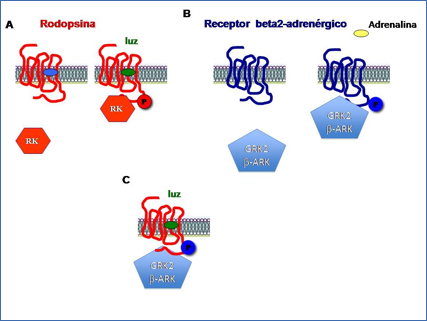
Figura 5.- Similitud entre los mecanismos de regulación por fosforilación de la rodopsina y del receptor beta-2 adrenérgico promovidos por la presencia de agonistas.
Se vislumbraba, por tanto, la emergencia de una "familia" de receptores para estímulos externos muy diversos, pero que conservaba unos rasgos estructurales, de funcionamiento y de regulación común: estaba naciendo lo que luego resultó ser la gran familia de receptores acoplados a proteínas G (GPCR), también llamados por sus características estructurales receptores de siete dominios transmembrana, o receptores "serpentina".
En los siguientes años, el clonaje por el laboratorio de Lefkowitz de diversos subtipos de receptores adrenérgicos (revisado en 23 y 24), y de un receptor muscarínico de acetil-colina por el grupo de Numa (27) corroboró la idea de la existencia de una gran familia de receptores relacionados estructural y funcionalmente.
Por otra parte, Lefkowitz y Kobilka realizaron experimentos con receptores quiméricos que combinaban secuencias de receptores alfa-2 y beta-2 adrenérgicos que fueron muy relevantes para identificar dominios intracelulares del receptor implicados en la interacción con subtipos de proteínas G específicos (28). También el grupo de Lefkowitz, con especial protagonismo de su colaborador J.L. Benovic, desarrolló a finales de la década de 1980 y en la década de 1990 la caracterización en detalle de los mecanismos de regulación de GPCRs por las proteínas GRKs y arrestinas, abriendo nuevos perspectivas sobre el funcionamiento y los mecanismos de señalización de esta familia de proteínas (29, 30).
5. Desvelando la estructura tridimensional de los receptores acoplados a proteínas G.
A pesar del inmenso progreso que suponían estos nuevos avances, persistían algunas preguntas clave: ¿cómo y dónde se unen los ligandos de GPCRs de forma específica? ¿cómo se transmite la señal y se promueve la activación de las proteínas G? La respuesta a estas preguntas requirió de nuevos experimentos bioquímicos y biofísicos y, muy particularmente, precisaba dilucidar la estructura cristalina de estos receptores.
Tras trasladarse a la Universidad de Stanford (California) en 1989, Brian Kobilka se propuso un reto que tardó casi 20 años en alcanzar: determinar la estructura en el espacio de esos receptores. Este proyecto presentaba algunos retos muy difíciles de resolver: los GPCRs son proteínas de membrana de poca abundancia relativa y son proteínas muy dinámicas, capaces de adoptar diversas conformaciones, así como altamente inestables en detergentes, presentando poca exposición de superficies hidrofílicas. Todo ello suponía un auténtico reto a la hora de intentar su cristalización.
Para hacer frente a estas dificultades el grupo de Brian Kobilka en colaboración con investigadores como Gebhard Schertler y Raymond Stevens desarrollaron diversas soluciones alternativas (31-33), que incluyeron el desarrollo de sistemas de expresión de alto rendimiento de receptores recombinantes en baculovirus, la utilización de nuevos detergentes, la co-cristalización de receptores con antagonistas o agonistas inversos capaces de estabilizar conformaciones específicas del receptor; el incremento del área hidrofílica de los receptores para facilitar la cristalización mediante la fusión con fragmentos tipo FAb o “nanobodies” o con T4 lisozima, la utilización de mutantes de receptores con mayor estabilidad…
Todo ello permitió publicar en el año 2007 la primera estructura del receptor beta2-adrenérgico unido al antagonista carazolol (31). Este avance facilitó también la comparación detallada con las estructuras tridimensionales de la rodopsina que, gracias a su mayor abundancia y facilidad de purificación, se habían obtenido por diversos autores particularmente Krzystztof Palczewski y Okada alrededor del año 2000 (34,35).
Estos progresos en la "ingeniería" de GPCRs y su cristalografía han permitido un avance acelerado en los últimos años en el conocimiento de nuevas estructuras. A finales del año 2012 se habían obtenido 16 estructuras de GPCRs, 9 de ellas publicadas en el propio año 2012 (revisado en referencia 36) . Entre estas estructuras se incluyen las de los receptores beta-1 adrenérgicos, muscarínicos tipo M2 y M3, el receptor H1 de histamina, el receptor D3 de dopamina, el receptor tipo A2a de adenosina, el receptor de esfingosina 1 fosfato S1P1, el receptor de quimioquinas CXCR4, tres subtipos de receptores de opiáceos (OPRK1, OPRM1, OPRD1), el receptor de nociceptina OPRL1, así como receptores de neurotensina y el receptor de proteasas PAR1 (36,37). Además, algunos de ellos han sido co-cristalizados en complejo con diferentes ligandos, lo que ha permitido investigar los cambios conformacionales relacionados con la unión de diversos compuestos químicos.
La obtención de estas diversas estructuras ha permitido conocer con precisión los dominios de receptores implicados en la interacción con sus ligandos específicos y las similitudes y diversidades estructurales presentes en los dominios extracelulares, transmembrana, e intracelular de los diversos GPCRs (revisado en detalle en las referencias 36 y 37).
La última frontera en este conocimiento de la estructura tridimensional de los receptores ha sido la identificación de los cambios estructurales que tienen lugar tras la activación del receptor y que conducen a la estimulación de las proteínas G. Para ello una vez mas ha sido decisiva la aportación de Brian Kobilka, publicada en dos artículos sucesivos en la revista Nature el 29 de septiembre del año 2011 (38,39).
En estos trabajos su grupo describió la cristalización de un receptor beta-2 adrenérgico activado por agonista en complejo con la proteína Gαs. Experimentos de una gran complejidad técnica, que incluyen la ingeniería de estas proteínas para favorecer la estabilización de ese complejo, han permitido identificar las superficies de interacción y los principales cambios estructurales que tienen lugar en las diversas proteínas del complejo (Figura 6).
En el caso del receptor beta-2 adrenérgico, el cambio conformacional más significativo incluye un movimiento hacia el exterior del extremo citoplásmico del segmento transmembrana 6 y una extensión en alfa-hélice del extremo citoplásmico del segmento transmembrana 5. Estos dos movimientos coordinados permiten la separación de los bucles intracelulares 2 y 3 de la estructura del receptor y la creación de un “bolsillo” hidrofóbico donde puede unirse la proteína Gαs (Figura 6). En concreto, la inserción en de la hélice α5 C-terminal de Gαs en esta hendidura hidrofóbica transitoria formada en el receptor activado por agonista permite a su vez cambios importantes en la estructura de la proteínas G, mediante la rotación de su dominio GTPasa y el remodelado de la región β6–α5, lo que facilitaría la liberación de GDP y el consiguiente paso de la proteína G a su estado activo.
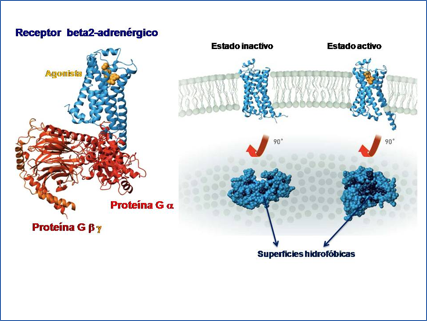
Figura 6.- Estructura tridimensional del receptor beta-2 adrenérgico y cambios promovidos tras su activación. Los trabajos de Kobilka han permitido cristalizar el receptor beta-2-adrenérgico activado por agonista en complejo con la proteína G e identificar las principales superficies de interacción entre estas proteínas. En el esquema de la derecha se muestran los principales cambios conformacionales promovidos por agonistas, que conducen a un aumento de las superficies hidrofóbicas que se ofrecen a las proteínas G en el interior de la célula (esquema modificado de www.nobel.org).
En definitiva, todos estos estudios están permitiendo obtener información muy relevante sobre las distintas conformaciones activas de los GPCRs y sobre cómo son capaces de transmitir información al interior de la célula. El concepto general que parece emerger es que la familia de receptores de 7 dominios transmembrana tendría una arquitectura general modular, compuesta por un módulo de unión de ligandos y otro módulo de señalización hacia el interior celular (Figura 7).
El módulo de unión de ligandos, formado por las bucles extracelulares del receptor y la parte más externa de sus dominios transmembrana, presenta la mayor diversidad entre los distintos receptores GPCR (lo que permitiría explicar su interacción específica con múltiples ligandos diferentes) y sufre cambios conformacionales menos acusados en presencia de agonistas (37). Por el contrario, el modulo de señalización hacía el interior de la célula, formado por la parte más interna de los dominios transmembrana y por los bucles intracelulares del receptor, presenta menor diversidad entre las subfamilias de GPCRs y sufre cambios conformaciones muy evidentes tras la llegada de los ligandos, permitiendo así la transmisión de la señal al interior de la célula (37). El hecho de que este último módulo sea más conservado es también coherente con el hecho de que 800 receptores para sustancias diferentes puedan converger en la interacción con las mismas proteínas G, o ser regulados por las mismas familias de quinasas GRKs y de arrestinas.
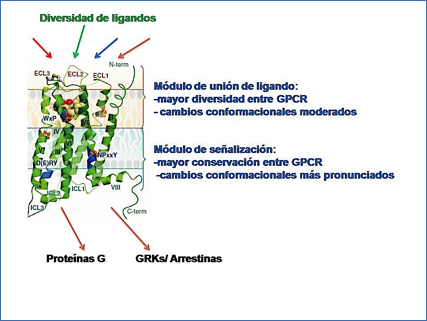
Figura 7.- Arquitectura modular de la familia de receptores acoplados a proteínas G Los GPCR se estructuran en dos módulos con distintos grados de divergencia de secuencia y de movilidad conformacional (esquema inspirado en la referencia 37).
6. Perspectivas futuras
La concesión del Nobel de Química a Lefkowitz y Kobilka completa justamente el reconocimiento que en años precedentes se había ido otorgando a los otros descubridores de pautas esenciales de este lenguaje de comunicación celular (Earl Sutherland, premio Nobel 1971 por identificar por primera vez el AMPc como segundo mensajero y Al Gilman y Martin Rodbell, premio Nobel 1994 por su descubrimiento de las proteínas G). Sin embargo, hay todavía muchos interrogantes abiertos en este campo.
Entre ellos, cabe destacar las nuevas vías de señalización de receptores de 7 dominios transmembrana consecuencia de su interacción con las proteínas GRKs y arrestinas. Estas proteínas no solamente actuarían como reguladores negativos del acoplamiento de los GPCR a las proteínas G (que es como fueron identificados), sino que presentan complejos interactomas que les permiten también participar en la propagación de la señal de estos receptores tras la llegada de un agonista (30,40). Esta divergencia de señalización de los receptores de 7 dominios transmembrana entre vías dependientes de proteínas G y otras cascadas dependientes de GRKs y arrestinas ha llevado también a la identificación de los que se ha denominado “biased ligands”, sustancias químicas que de manera preferente inducen la interacción del receptor bien con proteínas G, bien con GRKs/arrestinas, lo que puede tener un gran interés para el desarrollo de nuevos fármacos. En este mismo sentido, el mejor conocimiento de los “bolsillos” de unión de ligandos a los GPCRs facilitará el diseño de nuevos compuestos químicos con mayor afinidad y/o selectividad (4,36,37).
Por otra parte, se está identificando la existencia de GPCRs codificados por diversos virus, que podrían tener un papel relevante en su capacidad patogénica y constituir potenciales dianas para su tratamiento.
Por último, la secuenciación del genoma humano ha puesto de manifiesto la presencia en nuestra información genética de múltiples miembros de la familia de receptores 7 dominios transmembrana sin mensajero fisiológico conocido (los denominados GPCRs huérfanos), lo que presenta el importante reto de identificar sus ligandos endógenos y sus funciones fisiológicas e implicaciones patológicas.
En definitiva, el camino abierto con extraordinaria clarividencia y perseverancia por Lefkowitz y Kobilka, seguido actualmente por muchísimos otros investigadores, permitirá seguir conociendo mejor las alteraciones de receptores en situaciones patológicas y avanzar en el diseño de nuevas estrategias terapéuticas. Hay aún mucho trabajo por hacer y muchas preguntas por contestar.
7. Referencias
1. Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3, 639-650. (2002)
2. Nordstrom, K. J.; M. Sallman Almen, et al. Independent HHsearch, Needleman-Wunsch-based, and motif analyses reveal the overall hierarchy for most of the G protein-coupled receptor families. Mol Biol Evol 28, 2471-2480 (2011).
3. Fredriksson, R; Schioth H. B. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol Pharmacol 67, 1414-1425. (2005).
4. Lagerstrom, M. C.; Schioth H. B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7, 339-357 (2008).
5. Sprang, S. R. G protein mechanisms: insights from structural analysis. Annu Rev Biochem 66, 639-678 (1997).
6. Sprang, S. R. G proteins, effectors and GAPs: structure and mechanism. Curr Opin Struct Biol 7, 849-856 (1997).
7. Overington, J. P.; Al-Lazikani, B. How many drug targets are there?. Nat Rev Drug Discov 5, 993-996 (2006).
8. Tyndall, J. D.; R. Sandilya. GPCR agonists and antagonists in the clinic. Med Chem 1, 405-421 (2005).
9. Langley, J. N. Observations on the physiological action of extracts of the supra-renal bodies. J Physiol 27, 237-256 (1901).
10. Dale, H. H. On some physiological actions of ergot. J Physiol 34, 163-206 (1906).
11. Ahlquist, R. P. Adrenergic receptors: a personal and practical view. Perspect Biol Med 17, 119-122 (1973).
12. Rall, T. W.; Sutherland, E. W. Formation of a cyclic adenine ribonucleotide by tissue particles. J Biol Chem 232, 1065-1076. (1958).
13. Walsh, D. A.: Perkins, J. P. et al. An adenosine 3',5'-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem 243, 3763-3765 (1968).
14. Rodbell, M.; Birnbaumer, L. et al. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action. J Biol Chem 246, 1877-1882 (1971).
15. Ross, E. M.; Gilman A. G. Resolution of some components of adenylate cyclase necessary for catalytic activity. J Biol Chem 252, 6966-6969 (1977).
16. Lefkowitz, R. J.; Mukherjee, C. et al. Stereospecific (3H)(minus)-alprenolol binding sites, beta-adrenergic receptors and adenylate cyclase. Biochem Biophys Res Commun 60, 703-709 (1974).
17. De Lean, A.; Stadel, J. M. et al. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem 255, 7108-7117 (1980).
18. Limbird, L. E.; Gill, D. M. et al. Agonist-promoted coupling of the beta-adrenergic receptor with the guanine nucleotide regulatory protein of the adenylate cyclase system. Proc Natl Acad Sci USA 77, 775-779 (1980).
19. Kent, R.A.; DeLean, A; Lefkowitz, R.J. A quantitative-analysis of beta-adrenergic receptors interactions: resolution of high and low affinity states of the receptor by computer modeling of ligand binding data. Mol. Pharm. 17, 14-23. (1979)
20. Caron, M. G. ; Lefkowitz, R. J. Solubilization and characterization of the beta-adrenergic receptor binding sites of frog erythrocytes. J Biol Chem 251, 2374-2384 (1976).
21. Shorr, R. G.; Lefkowitz, R. J. et al. Purification of the beta-adrenergic receptor. Identification of the hormone binding subunit. J Biol Chem 256, 5820-5826 (1981).
22. Benovic, J. L.; Shorr, R. G. et al. The mammalian beta 2-adrenergic receptor: purification and characterization. Biochemistry 23(20): 4510-4518 (1984).
23. Lefkowitz, R. J. Seven transmembrane receptors: a brief personal retrospective. Biochim Biophys Acta 1768 748-755 (2007).
24. Lefkowitz, R. J. Historical review: a brief history and personal retrospective of seven-transmembrane receptors." Trends Pharmacol Sci 25(8): 413-422 (2004).
25. Dixon, R. A.; B. K. Kobilka, et al. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature 321, 75-79 (1986).
26. Benovic, J. L.; F. Mayor, Jr., et al. Light-dependent phosphorylation of rhodopsin by beta-adrenergic receptor kinase. Nature 321, 869-872 (1986).
27. Kubo, T.; Fukuda, K. et al. Cloning, sequencing and expression of complementary DNA encoding the muscarinic acetylcholine receptor. Nature 323, 411-416 (1986).
28. Kobilka, B. K.; Kobilka, T. S. et al. Chimeric alpha 2-,beta 2-adrenergic receptors: delineation of domains involved in effector coupling and ligand binding specificity. Science 240, 1310-1316 (1988).
29. Pitcher, J.A; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu Rev Biochem 67, 653-792 (1998).
30. Lefkowitz ,R.J.; Shenoy, S.K. Transduction of receptor signals by beta-arrestins. Science 308, 512-517 (2005)
31. Rasmussen, S. G.; Choi, H. J. et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450, 383-387 (2007).
32. Cherezov, V.; Rosenbaum, D. M. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258-1265 (2007).
33. Rosenbaum, D. M.; Cherezov, V. et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318, 1266-1273 (2007).
34. Palczewski, K.; Kumasaka, T. et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739-745 (2000).
35. Okada, T.; Fujiyoshi, Y. et al. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci USA 99, 5982-5987. (2002).
36. Katritch, V.; Cherezov, V. et al. Structure-function of the g protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol 53, 531-556 (2013).
37. Katritch, V.; Cherezov, V. et al. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 33, 17-27 (2012).
38. Rasmussen, S.G.; DeVree, B.T. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549-555 (2011).
39. Chung, K. Y.; Rasmussen, S. G. et al. Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature 477, 611-615 (2011).
40. Penela, P., Murga, C. et al. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol 160, 821-832 (2010).
El Premio Nobel en Fisiología o Medicina 2012: Rebobinando la película genética del desarrollo
Juan Ramón Lacadena
Académico de Número de la Real Academia Nacional de Farmacia.
e-mail: edicion@ranf.com
Recibido el 18 de febrero de 2013 An. Real Acad. Farm. Vol 79, Nº 1 (2013), pag. 151-171.
RESUMEN
En el presente trabajo se glosan las investigaciones sobre reprogramación celular llevadas a cabo mediante transferencia nuclear de células somáticas y la obtención de células troncales pluripotentes inducidas (iPS). |
Palabras clave: Reprogramación celular; Transferencia nuclear; Células troncales pluripotentes inducidas; células iPS.
ABSTRACT
Rewinding the genetic film of development
Investigations on cell reprogramming carried out by cell nuclear transfer and the induction of somatic pluripotent stem cells (iPS) in mammals awarded the Nobel Prize in Phisiology or Medicine 2012 are analyzed. |
Keywords: Cell reprogramming; Nuclear transfer; Induced pluripotent stem cells; iPS cells.
1. Introducción
El 8 de octubre de 2012 se hacía público que la Asamblea Nobel del Instituto Karolinska había concedido el Premio Nobel en Fisiología o Medicina 2012 a los Dres. Sir John B. Gurdon (Gurdon Institute, Universidad de Cambridge, UK) y Shinya Yamanaka (Universidad de Kyoto, Japón) “por el descubrimiento de que células maduras [diferenciadas] pueden ser reprogramadas para convertirse en pluripotentes”. Permítaseme recordar aquí que en el discurso inaugural del año 2011 que tuve el honor de pronunciar en esta Real Academia recogí las siguientes palabras en el apartado correspondiente a las células troncales pluripotentes inducidas: “El hecho de que Yamanaka junto con Gurdon recibieran en 2009 el Premio Albert Lasker de Investigación Básica en Medicina puede ser una anticipación de que, antes o después, recibirán el merecido premio Nobel”. No tardó dos años en cumplirse mi profecía.
En la concesión de un premio Nobel puede premiarse una investigación pionera en un campo científico o una investigación posterior basada en el cambio de paradigma que supuso aquella. En el caso que hoy nos ocupa, la Asamblea Nobel ha tenido el acierto de premiar ambas cosas: por un lado, a Sir John B.Gurdon que hace 50 años, en 1962, demostró la posibilidad de que la información genética contenida en el núcleo de células diferenciadas de un anfibio pudiera ser reprogramada para reiniciar un proceso de desarrollo completo y, por otro lado, a Shinya Yamanaka que 44 años más tarde encontró las claves genéticas para inducir la reprogramación celular en mamíferos como el ratón y el ser humano
2. CONCEPTO GENÉTICO DE DESARROLLO
El desarrollo se puede definir como un “proceso regulado –es decir, bajo control genético– de crecimiento y diferenciación resultante de la interacción núcleo-citoplásmica, del ambiente celular interno y del medio externo mediante el cual se produce la formación del individuo adulto a partir de una célula inicial única: el cigoto originado por la fecundación de los gametos”. El cigoto reúne la información genética necesaria (aunque a veces no suficiente) para programar la formación del nuevo ser, de manera que, de no mediar alteraciones de cualquier tipo que interfieran con el proceso, a partir del momento en que empiece a funcionar el primer gen en dicha célula, la programación genética conducirá inexorablemente a la formación del individuo adulto. El proceso de desarrollo es, por tanto, una secuencia programada de cambios fenotípicos controlados espacial y temporalmente que constituyen el ciclo vital del organismo (1).
En el proceso global de desarrollo cabe distinguir los siguientes fenómenos o componentes del desarrollo:
· La replicación genética (ADN) y la proliferación celular, que producen el crecimiento;
· La diferenciación celular o citodiferenciación, fenómeno por el cual células que tienen un origen común y, por tanto, son genéticamente idénticas, divergen en su estructura y/o función, dando lugar a líneas celulares morfológicamente y/o fisiológicamente diferentes;
· La histogénesis, como resultado de la agregación de células diferenciadas para constituir un tejido con función especializada;
· La organogénesis, como consecuencia de la asociación de tejidos, dando como resultado final la forma del individuo (morfogénesis);
· Por último, podría considerarse el comportamiento como una expresión multidimensional del desarrollo.
En el contexto del Premio Nobel que conmemoramos, solamente haremos referencia a la diferenciación celular. La citodiferenciación es debida a una actividad génica diferencial determinada por causas ambientales intra o extra celulares aunque en algunas ocasiones pueda ser producida por o ir acompañada de modificaciones cromosómicas estables (numéricas, estructurales o fisiológicas) de la dotación cromosómica de las células. La cuestión fundamental que se plantea es si las células diferenciadas conservan la capacidad (multipotencia o pluripotencia) de originar un tejido u órgano diferente al que estaban programadas o a un organismo completo (totipotencia), comportándose como un verdadero cigoto si las condiciones experimentales las indujeran a ello.
3. REPROGRAMACIÓN NUCLEAR
Desde el punto de vista genético, por reprogramación nuclear se entiende el cambio en la expresión génica de una clase de célula a otro tipo de célula no relacionada con ella. Como señalaban Gurdon y Melton (2), las primeras evidencias experimentales sobre reprogramación se obtuvieron en las décadas de los 50 y 60 del siglo pasado con los trabajos sobre clonación en anfibios. A partir de ahí habría que tener en cuenta las técnicas de clonación por transferencia nuclear de células somáticas en mamíferos (SCNT, somatic cell nuclear transfer), la fusión celular, las células troncales embrionarias y adultas, la inducción de pluripotencia por expresión génica ectópica (células iPS, induced pluripotent stem cells) y la reprogramación directa que permite transformar un tipo de célula diferenciada en otra célula diferenciada (transdiferenciación) sin pasar por la fase intermedia equivalente de una célula pluripotente. Lo mismo que en la Edad Media los alquimistas trataban de transmutar en oro a otros metales, en la actualidad estamos viviendo una nueva clase de alquimia -la alquimia celular (3)- que convierte una célula en otro tipo de célula.
El Premio Nobel en Fisiología o Medicina 2012 que hoy conmemoramos tiene que ver con la clonación por transferencia de núcleos (Gurdon) y con la inducción de células troncales pluripotentes (Yamanaka).
4. LA CLONACIÓN POR TRANSFERENCIA DE NÚCLEOS
4.1. Anfibios
En la década de los cincuenta del siglo pasado, Briggs y King (4) trasplantaron núcleos de células de blástula, gástrula, néurula y renacuajo de Rana pipiens a citoplasmas de óvulos sin fecundar que habían sido enucleados mediante micro manipulaciones para comprobar si tales núcleos eran capaces de dar marcha atrás en su proceso informativo y volver a dar un desarrollo normal. Los resultados obtenidos mostraron que al trasplantar núcleos del estadio de blástula se obtenía un desarrollo normal, mientras que al trasplantar los núcleos de células de gástrula, néurula o renacuajo disminuía de forma progresiva la capacidad de desarrollo. La conclusión evidente era que cuanto más diferenciadas estaban las células donantes de los núcleos tenían menos capacidad de desarrollo total (totipotencia).
Sin embargo, diez años después del primer experimento de Briggs y King, en 1962 John B. Gurdon (5) hizo un experimento que le ha valido el Premio Nobel sesenta años más tarde, porque su investigación cambió la idea de que la diferenciación celular era un proceso irreversible, sentando las bases para el desarrollo posterior de las técnicas de reprogramación nuclear, tanto en la obtención de mamíferos clónicos como en la obtención de células troncales, de especial importancia en la Biomedicina.
Los experimentos de Gurdon consistieron en transferir el núcleo de una célula diferenciada (célula ciliada epitelial de intestino) de renacuajo del sapo con garras africano (Xenopus laevis) al citoplasma de un óvulo cuyo núcleo había sido destruido mediante radiación ultravioleta, obteniendo un sapo macho y otro hembra normales, aunque con una frecuencia pequeña (1%).
Como comprobación experimental de que la técnica de transferencia nuclear había sido correcta, Gurdon utilizó como cepa donadora del núcleo un mutante nucleolar obtenido por Fischberg (6), en cuyo laboratorio había trabajado con anterioridad, que mostraba en el núcleo interfásico un solo nucleolo en lugar de dos que tenía la cepa receptora normal. Diez años más tarde, Kobel y colaboradores (7) obtuvieron un sapo hembra fértil transfiriendo núcleos de células no ciliadas de epidermis de renacuajo, ratificando así las experiencias de Gurdon.
A pesar de la evidencia experimental aportada por Gurdon y la corroboración por Kobel y colaboradores, sin embargo la clonación en anfibios por transferencia de núcleos de células diferenciadas fue recibida con cierto escepticismo por parte de la comunidad científica (8) porque, como señala la propia Institución Nobel, el descubrimiento de Gurdon “hizo añicos el dogma de que la diferenciación celular sólo podía ser un proceso unidireccional” (9).
De hecho, la idea científica vigente entonces estaba muy enraizada con el modelo de canalización del desarrollo propuesto por Waddington en 1957 (10) en la década anterior en el que comparaba el proceso de desarrollo con un paisaje epigenético de montañas y valles en el que las células indiferenciadas están en las cumbres de las montañas y en el proceso de diferenciación entran en los valles de forma que ya no podrán volver al estado diferenciado que representan las cumbres.
Otro ejemplo muy gráfico que solía utilizar yo en mis clases de Genética era el de los cambios de vía de una estación de tren donde se clasificaban los vagones llevándolos a las vías muertas del la diferenciación.
4.2. Mamíferos
Si en anfibios había sido posible la clonación por transferencia de núcleos, ¿por qué no podía ser posible también en mamíferos? Sin embargo, las investigaciones realizadas inicialmente en ratones dieron resultados negativos, dando pie a que se llegara a decir en palabras del mayor exponente en el campo de la investigación genética embriológica en ratón de aquella época que “la clonación por transferencia de núcleos en mamíferos es biológicamente imposible” (11).
Es obvio que términos como imposible, siempre o nunca no pueden ser usados en cuestiones científicas. De hecho, trece años más tarde, en 1997, la comunidad científica y la sociedad conocieron con perplejidad que en el Instituto Roslin de Edimburgo había nacido la oveja Dolly originada por transferencia del núcleo de una célula de glándula mamaria de una oveja adulta a un ovocito enucleado en una investigación dirigida por el Dr. Ian Wilmut (12). Este trabajo del grupo de Wilmut abrió las puertas a la clonación por transferencia de núcleos en mamíferos; así, hasta la fecha de hoy, se han obtenido animales clónicos en otras muchas especies: ratón, vaca, mono rhesus, cabra, cerdo, gato, conejo, carnero “bateng”, mulo, caballo, rata, ciervo, perro, lobo, camello, toro de lidia, coyote. Cuando se conoció la noticia del nacimiento de la oveja Dolly, un periódico hacía un comentario con el titular “hoy la oveja, mañana el pastor” planteando el problema ético que supondría obtener humanos clónicos.
Como ha ocurrido en otras ocasiones, en el camino de la concesión de este Premio Nobel en Fisiología o Medicina 2012 ha habido algún damnificado: me refiero al Dr. Ian Wilmut (Roslin Institute, Universidad de Edimburgo, UK) y a su colaborador el Dr. Keith Kampbell −fallecido unos días antes de la concesión del premio por lo que no hubiera podido recibir el galardón según las normas de la institución Nobel− que muy bien podría haber compartido el premio con el Dr. Gurdon reconociendo su mérito por la clonación de la oveja Dolly que abrió las puertas a la clonación en mamíferos.
5. CÉLULAS TRONCALES PLURIPOTENTES INDUCIDAS (IPS) (13)
5.1. Células troncales
La terapia celular, basada en la transferencia de células o tejidos a los tejidos u órganos dañados, es una de las grandes esperanzas de la Medicina Regenerativa del futuro. El establecimiento de cultivos celulares de tejidos humanos en el laboratorio es a veces muy difícil. Por ello, desde el punto de vista clínico es innegable el avance que supondría la posibilidad de poner a punto técnicas que permitieran obtener cualquier tipo de cultivos de tejidos y, acaso en un futuro más lejano, de órganos. En este contexto, no cabe duda que el uso de las células troncales. puede resultar fundamental.
Por célula troncal se entiende cualquier célula indiferenciada que tiene la doble capacidad de dividirse de forma ilimitada y, en un cierto momento, diferenciarse dando lugar a diferentes tipos de células especializadas. De acuerdo con esta segunda capacidad, las células troncales pueden ser totipotentes, pluripotentes y multipotentes en razón a su mayor o menor versatilidad o potencialidad, tal como se definen a continuación:
· Célula totipotente: Célula troncal que tiene la capacidad de diferenciarse en el embrión y en tejidos y membranas extraembriónicas. Las células totipotentes contribuyen a todos los tipos celulares de un organismo adulto. La totipotencia es la capacidad funcional de una célula de dar lugar a un individuo completo tras un proceso de desarrollo normal. Las células totipotentes de un embrión muy temprano tienen la capacidad de diferenciarse en membranas y tejidos extraembriónicos, en el embrión y en todos los tejidos y órganos postembriónicos. En el embrión humano, parece ser que solamente son totipotentes los blastómeros hasta el estadio de mórula de 16 células.
· Célula pluripotente: Célula troncal presente en los estadios tempranos de desarrollo embrionario (blastocisto) que puede generar todos los tipos de células en el feto y en el adulto y es capaz de autorrenovación. Las células pluripotentes, sin embargo, no son capaces de desarrollarse en un organismo completo. La pluripotencia es la capacidad funcional de una célula de dar lugar a varios linajes celulares o tejidos diferentes. Las células troncales embrionarias (ES) presentes en la masa celular interna (MCI) del blastocisto humano son pluripotentes, pero no totipotentes; es decir, pueden originar distintos tejidos u órganos pero no dar lugar al desarrollo completo de un embrión porque no pueden producir las membranas y tejidos extraembriónicos necesarios para el proceso de gestación. No obstante, podría ocurrir que una célula pluripotente de la masa celular interna se convirtiera en totipotente.
· Célula multipotente: Célula troncal presente en los tejidos u órganos adultos que tiene una capacidad limitada de reactivar su programa genético como respuesta a determinados estímulos que le permiten dar lugar a algunos, pero no todos, los linajes celulares diferenciados. La multipotencia es la capacidad funcional de una célula de dar lugar a alguno, pero no todos, los linajes celulares. Algunas células troncales presentes en tejidos u órganos adultos son multipotentes. A veces se utiliza el término plasticidad como equivalente a multipotencia.
Hay varias clases de células troncales (embrionarias, adultas, pluripotentes inducidas) cuya eficacia en el establecimiento de cultivos de tejidos en el laboratorio y sus valoraciones éticas y jurídicas son diferentes. Aunque los tres tipos de células troncales pueden tener una aplicación clínica, sin embargo las células iPS son especialmente indicadas en la terapia celular de la Medicina Regenerativa porque al tratarse de una transferencia autóloga se evita el rechace inmunológico. En el presente contexto únicamente se hará referencia a las células troncales pluripotentes inducidas (células iPS).
5.2. Células roncales pluripotentes inducidas (iPS), una esperanza ética para el futuro (14)
Células iPS de ratón
Desde el año 2003, el laboratorio de Yamanaka (15) estaba interesado en el estudio de los factores necesarios para mantener la pluripotencia de las células troncales embrionarias, identificando el gen Nanog (16) a la vez que lo hacía el grupo de Austin Smith (17). A partir de entonces decidió investigar la posibilidad de inducir la pluripotencia en células somáticas. Conociendo el trabajo de Tada y colaboradores (18) del año 2001 en el que demostraron que la fusión de células somáticas con células ES induce la pluripotencia del núcleo somático, Yamanaka seleccionó un conjunto de 24 factores de transcripción como posibles candidatos para inducir la pluripotencia en células somáticas, descartando uno a uno los factores que no parecían capacitados para ello.
Finalmente, en 2006 Yamanaka (19), logró la reprogramación de células somáticas de ratón utilizando solamente cuatro genes reguladores de la transcripción (Oct3/4, Sox2, c-Myc y Klf4) que fueron capaces de convertir fibroblastos embriónicos de ratón en células troncales pluripotentes, por lo que se le puede acreditar la paternidad de la técnica (20) que fue ratificada por el grupo de investigación de Rudolf Jaenisch (21). Posteriormente, Yamanaka refinó la técnica original (22). En 2009, María Blasco y colaboradores demostraron que es necesaria la presencia de la actividad telomerasa en la obtención de células iPS en ratón (23).
Pensando en una futura aplicación clínica de estas técnicas hay que señalar dos inconvenientes: en primer lugar, la utilización de un retrovirus como vector para introducir los genes reguladores que reprograman las células somáticas y, en segundo lugar, en el caso de Yamanaka, la utilización del proto-oncogén c-Myc, lo cual puede suponer un obstáculo para lograr la autorización para llevar a cabo la investigación clínica. De hecho, un 20% de los ratones implantados con células iPS desarrollaron teratomas cancerosos. Más tarde, en 2011, Yamanaka y colaboradores (24) obtenían células iPS a partir de fibroblastos de ratón y humanos sustituyendo el factor c-Myc por el factor de transcripción Glis1 (de la familia de zinc finger 1), evitando el posible efecto dañino del proto-oncogén c-Myc. El factor Glis 1 promueve múltiples vías de reprogramación, tales como las Myc, Nanog, Lin28, Wnt, Essrb y la transición mesenquima-epitelial.
Para obviar estas dificultades clínicas se han introducido algunas mejoras técnicas: por ejemplo, Yamanaka y colaboradores (25) han obtenido las células iPS sin utilizar vectores virales en ratones. Para ello utilizaron la transfección repetida de dos plásmidos de expresión en fibroblastos de embriones, uno era portador de los ADNc (ADN complementario) de Oct3/4, Sox2 y Klf4 y el otro del de c-Myc. En las células iPS obtenidas no se produjo la integración del plásmido en el genoma, evitando la formación de teratomas.
Por su parte, Jaenisch y colaboradores (26), con objeto de reducir el número de partículas virales necesarias para la reprogramación y por tanto disminuir el peligro de la mutagénesis inducida por la integración de ADN viral en el genoma, lograron que los cuatro factores de reprogramación Oct4, Sox2, Klf4 y c-Myc pudieran ser expresados a partir de un único promotor viral, generando células iPS tanto a partir de células embrionarias o adultas de ratón como a partir de queratinocitos humanos.
Por otro lado, Melton y colaboradores (27) obtuvieron células iPS a partir de fibroblastos humanos utilizando solamente los factores Oct4 y Sox2, evitando la presencia de los proto-oncogenes Klf4 y c-Myc y sus posibles efectos dañinos. Incluso, más tarde, Schöler y colaboradores (28) demostraron que la presencia única del gen Oct4 bastaba para obtener células iPS (que denominan 1FiPS) a partir de células troncales neuronales de ratón adulto.
Un nuevo avance en el tema de la células iPS se produjo en abril de 2009 cuando Ding y colaboradores (29) lograron transformar células somáticas de ratón en células troncales pluripotentes inducidas (iPS) sin introducir en las células información genética alguna sino, simplemente, poniendo a las células en presencia de determinadas proteínas.
Células iPS humanas
El paso a la obtención de células troncales pluripotentes inducidas en la especie humana la realizaron casi simultáneamente el grupo de Yamanaka y el de Thomson. En 2007, Thomson y colaboradores (30) lograron reprogramar células somáticas adultas humanas (procedentes de prepucio de recién nacido y de piel de feto) y convertirlas en células troncales pluripotentes, introduciendo en ellas mediante un vector viral cuatro genes (Oct4, Sox2, Nanog y Lin28) que regulan la transcripción (factores de transcripción). Estas células troncales pluripotentes inducidas (células iPS, induced pluripotent stem cells) tienen cariotipo normal, expresan actividad telomerasa, expresan marcadores celulares de superficie y genes que caracterizan a las células troncales embrionarias (ES) y mantienen el potencial de desarrollo para diferenciarse en células de las tres capas germinales primarias. La eficacia de la técnica es de una célula iPS obtenida por cada 10.000 células tratadas que, en términos prácticos, es muy alta.
La publicación del trabajo del grupo de Thomson fue simultánea con la del grupo de Shinya Yamanaka (31) quienes utilizando como vector un retrovirus introdujeron en células somáticas humanas (células de la piel de la cara de una mujer de 36 años y de tejido conectivo sinovial de un varón de 69 años) cuatro genes reguladores de la transcripción (Oct3/4, Sox2, c-Myc y Klf4) con una eficacia de una célula troncal pluripotente inducida (iPS) obtenida por cada 5.000 células de piel utilizadas en el tratamiento. Por su parte, Izpisúa Belmonte y su grupo (32) mostraron que la utilización de queratinocitos de cabello humano resultaba cien veces más eficaz y era el doble de rápido que cuando se utilizaban fibroblastos.
Como se ha indicado anteriormente, Melton y colaboradores obtuvieron en 2008 células iPS a partir de fibroblastos humanos utilizando solamente los factores Oct4 y Sox2, evitando la presencia de los proto-oncogenes Klf4 y c-Myc y sus posibles efectos dañinos.
En noviembre de 2010, Rossi y colaboradores (33) obtuvieron células iPS a partir de varios tipos de células humanas mediante ARNm sintético modificado (RiPS cells), evitando los peligros de los métodos integrativos de ADN (vectores virales) y logrando superar la eficacia de otros métodos diseñados anteriormente (por ejemplo, uso de proteínas). Ellos sintetizaron ARN modificado correspondiente a los cuatro factores de transcripción canónicos utilizados inicialmente por Yamanaka: Klf4, c-Myc, Oct4 y Sox2 y en un experimento posterior añadieron el ARN modificado correspondiente a un quinto factor (Lin28 de Thomson). Los métodos utilizados no son mutagénicos y son altamente controlables. Además, la utilización de ARN modificado plantea la posibilidad de utilizar esta nueva tecnología para dirigir la diferenciación de las células iPS obtenidas (RiPS) en el tipo celular deseado. Por ejemplo, los mismos autores, utilizando ARN sintético modificado que codificaba para el factor de transcripción miogénico MYOD, obtuvieron células miogénicas que originaban miotubos formados por miogenina y miosina.
Aplicación terapéutica en modelo experimental de ratón
En diciembre de 2007, el grupo de Jaenisch hizo público en la revista Science el éxito de la aplicación en ratones de la técnica de Yamanaka para el tratamiento de la anemia falciforme humana en un modelo de ratón utilizando células pluripotentes inducidas (iPS) mediante reprogramación de células de la piel (34). Posteriormente, en 2009, Ward, Ma y colaboradores (35) lograron corregir también en ratones la hemofilia de tipo A (producida por mutación del factor VIII) de manera que al inducir un corte en la cola de ratones inyectados en el hígado con células iPS no sufrían daños importantes mientras que los ratones control no inyectados morían en pocas horas. Al año siguiente, en 2010, el mismo grupo lograba revertir la hiperglicemia en ratones diabéticos de tipos 1 y 2 mediante la obtención y transferencia de células beta pancreáticas a partir de células iPS (36).
En 2010, Yamanaka, Okano y colaboradores (37) obtuvieron neuroesferas “seguras” (que no inducen tumorogénesis) derivadas de células iPS que originaban in vitro neuronas, astrocitos y oligodendrocitos de ratón electrofisiológicamente funcionales. Además, cuando dichas neuroesferas “seguras” eran trasplantadas a la médula espinal de un ratón 9 días después de haberle producido el daño, se diferenciaban en los tres linajes celulares neurales sin formar teratomas ni tumores, participando en la remielización e inducción del recrecimiento axonal de las fibras serotonérgicas, contribuyendo a la recuperación de la función locomotora.
Aplicación terapéutica en humanos
Estas investigaciones suponen un paso adelante esperanzador en la posible utilización de células iPS en la terapia celular humana del futuro, obviando los problemas éticos de la manipulación de embriones.
La aplicación terapéutica de las células iPS podría plantearse en tratamiento clínicos in vivo sobre los pacientes que necesitarán disponer de las garantías suficientes que eviten efectos secundarios nocivos como puede ser la producción de tumores o en experimentos de laboratorio in vitro según el modelo de “enfermedades en placa petri” que permitan conocer los procesos de la enfermedad o realizar pruebas toxicológicas para el desarrollo de nuevos fármacos.
Efectivamente, en 2008, Eggan y colaboradores (38) lograron mediante la técnica de inducción de células iPS generar in vitro a partir de fibroblastos de piel células nerviosas motoras en un paciente de 82 años que padecía esclerosis lateral amiotrófica (ELA), que son precisamente las células dañadas por la enfermedad. La técnica consistió en introducir en los fibroblastos los genes Klf4, Sox2, Oct4 y c-Myc utilizando como vector un retrovirus. También Park y colaboradores indujeron la obtención de células iPS en casos de distrofia muscular y de la enfermedad de Huntington (39).
Posteriormente, en 2009, Svendsen y colaboradores (40) obtuvieron células iPS a partir de fibroblastos de piel de un niño afecto de atrofia muscular espinal (AME), enfermedad autosómica recesiva que suele manifestarse a partir de los 6 meses de edad y que produce la muerte del paciente en torno a los dos años. Las células iPS obtenidas generaban neuronas motoras defectuosas de manera que, como dicen los autores del trabajo, se pueden estudiar comparativamente con las células nerviosas homólogas producidas por la madre fenotípicamente sana del niño enfermo y poder así estudiar los mecanismos de la enfermedad. En la técnica se utilizaron los genes Oct4, Sox2, Nanog y Lin28 que fueron introducidos en los fibroblastos utilizando como vector un lentivirus.
Las células iPS específicas del síndrome de Rett muestran una disminución en la densidad de espinas después de la diferenciación neuronal (41).
La diferenciación de hepatocitos a partir de células iPS en pacientes con deficiencia para la α1-antitripsina produce una acumulación elevada de lípidos y glicógeno (42).
En 2011, Izpisúa y colaboradores (43) obtuvieron céulas iPS sanas a partir de fibroblastos de piel de niños que padecían la enfermedad de Hunchinson-Gilford de envejecimiento prematuro (progeria) y que al ser rediferenciadas a células musculares lisas volvían a manifestar las características de la enfermedad (acumulación de progerina y desorganización de la lamina nuclear). Sus investigaciones pueden permitir estudiar las causas del envejecimiento in vitro.
Recientemente, también de han utilizado las células iPS en enfermedades de manifestación tardía como la ataxia espinocerebelar (44), la enfermedad de Alzheimer (45), la enfermedad de Huntington (46) y la enfermedad de Parkinson: en 2012, Izpisúa y colaboradores (47) obtuvieron células iPS a partir de células de la piel de pacientes con la enfermedad de Parkinson comprobando que las células iPS presentaban una anomalía en la membrana nuclear semejante a la que presentaban las células de una muestra post mortem del cerebro de pacientes que habían padecido dicha enfermedad. También se han iniciado estudios con enfermedades complejas como la esquizofrenia (48).
A modo de resumen, en el Cuadro 1 se resumen las investigaciones más importantes descritas anteriormente indicando los autores y las fechas correspondientes:
Cuadro 1.- Células troncales pluripotentes inducidas (iPS).
Células iPS en ratón |
Yamanaka (2006, 2008); Jaenisch (2007, 2009); Schöler (2009); Blasco (2009); Ding (2009) |
Células iPS humanas |
Yamanaka (2007, 2011); Thomson (2007); Park (2008); Jaenisch (2008, 2009); Lowry (2008); Izpisúa Belmonte (2008); Melton (2008); Rossi (2010); Gage (2011) |
Aplicaciones terapéuticas en ratón como modelo experimental |
Anemia falciforme HbS (Jaenisch, 2007); hemofilia A (Ward y Ma, 2009); diabetes (Ward y Ma, 2010); lesiones en médula espinal (Yamanaka y Okano, 2010) |
Aplicaciones terapéuticas en humanos |
Esclerosis lateral amiotrófica, ELA (Eggan, 2008); distrofia muscular y enfermedad de Huntington (Park, 2008 y The HD iPSC Consortium, 2012); atrofia muscular espinal, AME (Svendsen, 2009); síndrome de Rett (Marchetto, 2010); deficiencia en α1-antitripsina (Rashid, 2010); ataxia espinocerebelar (Koch, 2011); progeria (Izpisúa Belmonte, 2011); esquizofrenia (Brennand, 2011); enfermedad de Parkinson (Izpisúa Belmonte, 2012); enfermedad de Alzheimer (Israel, 2012) |
La importancia del descubrimiento de la posibilidad de obtener células troncales pluripotentes inducidas (células iPS) por reprogramación celular de células somáticas adultas mereció ser seleccionado por la revista Science (49) como el segundo de los diez descubrimientos científicos más importantes del año 2007 y como el más importante del año 2008. Consideraba la revista que la obtención de células iPS es el logro de “una hazaña largamente buscada de alquimia celular”: así como los antiguos alquimistas buscaban convertir metales vulgares en oro, los científicos actuales han logrado convertir células humanas diferenciadas en células iPS, el equivalente biológico del oro.
Al igual que le ha sucedido al Dr. Ian Wilmut con la clonación en relación con el galardón Nobel concedido al Dr. Gurdon que he comentado anteriormente, el Dr. James A. Thomson ha experimentado una situación análoga por partida doble. En efecto, la historia de la células troncales pluripotentes embrionarias empezó en 1981 cuando Sir Martin J.
Evans −galardonado con el Premio Nobel en Fisiología o Medicina en 2007− logró aislarlas y cultivarlas a partir de embriones de ratón en fase de blastocisto (50). Años más tarde, en 1998, el grupo dirigido por James A. Thomson, de la Universidad de Wisconsin-Madison, consiguió aislar y mantener en cultivo células troncales embrionarias a partir de blastocistos humanos (51), continuando de alguna manera lo que Evans había logrado en ratones en 1981 y abriendo un nuevo campo científico en la Medicina. Este año por segunda vez se ve privado el Dr. Thomson del galardón Nobel a pesar de haber obtenido casi simultáneamente con el Dr. Yamanaka las células troncales pluripotentes inducidas humanas. Como es sabido, la normativa de la Institución Nobel prohíbe dar el mismo premio a más de tres personas, de no tratarse de un colectivo u organización.
Los intentos de abrir las puertas a la clonación humana con fines terapéuticos (obtención de embriones somáticos por transferencia nuclear de células del propio paciente) puede que resulten innecesarios si llega a hacerse una realidad clínica la reprogramación de células somáticas adultas utilizando las técnicas de Yamanaka, Thomson y Jaenisch antes descritas. En este contexto cabe señalar que el Dr. Ian Wilmut, padre científico de la oveja Dolly, anunció que abandonaba la investigación en clonación terapéutica humana para pasarse a la utilización de la técnica de reprogramación celular de Yamanaka. También José B. Cibelli, uno de los pioneros de la clonación humana (52), se ha manifestado a favor de la nueva técnica de reprogramación mientras que otros científicos siguen aferrándose a la investigación con células troncales embrionarias como a un clavo ardiendo.
Los defensores de continuar investigando con células troncales embrionarias humanas defienden su posición, argumentando que si no hubiera sido por estas investigaciones no se hubiera llegado a conocer el papel de los factores de transcripción Oct3/4, Sox2, c-Myc, Klf4, Nanog y Lin28 en el proceso de reprogramación celular de células somáticas adultas.
En el presente contexto, es importante señalar que, según datos de los NIH de los Estados Unidos (53), en diciembre de 2012 se habían registrado en todo el mundo 4118 investigaciones clínicas con células troncales adultas (AS), 24 con células troncales embrionarias (ES) y 19 con células troncales pluripotentes inducidas (iPS) (14 en Estados Unidos, 3 en Israel, 1 en Francia y 1 en Irán). Desde el punto de vista ético resulta interesante resaltar que la comunidad científica parece dispuesta a evitar la utilización de células troncales de embriones humanos (0,6%); incluso, en Estados Unidos hay 14 investigaciones clínicas con células iPS frente a 8 con células ES.
6. REPROGRAMACIÓN DIRECTA
En la década de los sesenta del siglo pasado, Ernst Hadorn (54) introdujo los conceptos de determinación y transdeterminación en sus estudios de genética del desarrollo en Drosophila. Según Hadorn, la diferenciación celular se inicia por el proceso de la determinación que programa la célula para su futuro comportamiento en el desarrollo.
En unos casos, la diferenciación celular es inmediata a su determinación mientras que en otros casos la determinación supone un estado celular reproducible propagado durante una fase indiferenciada más o menos larga hasta que en un momento dado esas células determinadas se diferencian. Se entiende por herencia celular a la propiedad individual de las células de mantener y transmitir su estado de determinación. Utilizando en su investigación discos imaginales de Drosophila (primordios celulares presentes en las larvas que durante la metamorfosis se transforman en estructuras específicas de la mosca adulta), Hadorn observó que ocasionalmente se podían producir cambios en la herencia celular, de manera que discos imaginales de una cierta estructura adulta (alas, patas, antenas, ojos, etc.) dan lugar a otra estructura. Al fenómeno que produce tales cambios en la herencia celular se le denomina transdeterminación.
La Institución Nobel (55) recordaba estos datos científicos de hace casi medio siglo por analogía con los procesos de reprogramación directa o transdiferenciación que en la actualidad están llevándose a cabo utilizando técnicas semejantes a las establecidas por Yamanaka.
Efectivamente, un nuevo paso conceptualmente diferente a la técnica de inducción de células troncales pluripotentes (iPS) de Yamanaka lo dio en 2008 el grupo de Douglas A. Melton (56) al reprogramar in vivo células adultas de ratón (células exocrinas del páncreas) transformándolas directamente (reprogramación directa o transdiferenciación) en células beta pancreáticas capaces de producir insulina (islotes de Langerhans). Las células obtenidas son indistinguibles de las células beta pancreáticas endógenas, tanto en tamaño como en su forma y estructura. Para ello, utilizaron como vector un adenovirus en el que se habían incorporado tres factores de transcripción (Ngn3, Pdx1 y MafA) que el grupo de Melton había identificado previamente como responsables de la diferenciación de las células beta pancreáticas. El experimento realizado con ratones in vivo mostró que las células beta obtenidas mejoraban sensiblemente la condición de hiperglicemia de los ratones diabéticos. No cabe duda que estos resultados son esperanzadores para tratar de curar en el futuro la enfermedad de la diabetes tipo 1 en humanos.
Más tarde, en 2010 y 2011, Wernig y colaboradores (57), partiendo de la hipótesis de que la expresión combinatoria de factores de transcripción específicos del linaje neural podría convertir directamente fibroblastos en neuronas, utilizaron un conjunto de 19 genes candidatos de los que solamente tres factores (Ascl3, Brn2 también denominado Pou3f2 y Myt1l) eran suficientes para convertir con rapidez y eficacia fibroblastos embrionarios y postnatales de ratón directamente en neuronas funcionales in vitro. Las células neuronales inducidas (iN) expresan múltiples proteínas específicas de neurona, generan potenciales de acción y forman sinapsis funcionales. Los autores señalaban que la generación de células iN a partir de linajes no neurales podría tener importantes implicaciones tanto en el estudio del desarrollo neural como en el diseño de modelos de enfermedades neurológicas y la Medicina regenerativa.
Bhatia y colaboradores (58) lograron la conversión directa de fibroblastos humanos en células progenitoras hematopoyéticas que daban lugar a linajes granulocíticos, monocíticos, megacariocíticos y eritroides. También en 2010, Ieda y colaboradores (59) transformaron in vitro fibroblastos en cardiomiocitos utilizando tres factores de transcripción y en 2012 dos grupos de investigación distintos (60,61) inducían in vivo la transformación directa de fibroblastos en cardiomiocitos de ratón utilizando los mismos tres factores de transcripción. Es importante resaltar que estas investigaciones muestran la posibilidad de lograr la transdiferenciación o reprogramación directa no sólo entre células dentro de una misma capa germinal, sino también entre células de capas germinales diferentes como es el caso de fibroblastos (mesodermo) a neuronas (ectodermo).
La nueva técnica de reprogramación directa se salta el paso intermedio de las células iPS por el que la célula tratada se revierte al estado de maduración de una célula para que sea pluripotente. Es evidente que se ha abierto un nuevo y esperanzador campo para la medicina regenerativa del futuro. Además, como en el caso de la utilización de las células troncales adultas (AS) y las células troncales pluripotentes inducidas (iPS), se obvian los problemas éticos que plantea la utilización de las células troncales pluripotentes embrionarias (ES).
7. EPIGÉNESIS
En el presente contexto, me parece interesante hacer un breve comentario sobre la epigénesis, teniendo en cuenta que su significado ha tenido algunas variaciones a lo largo del tiempo. La Genética del Desarrollo estudia los procesos genéticos que controlan el paso de cigoto a adulto. Las concepciones clásicas de preformación (Hartsoecker, 1695) y epigénesis (Wolff, 1759) encuentran una base genética en su sentido conceptual por cuanto, por un lado, el cigoto reúne ya en forma de ADN lo que ha de ser el nuevo individuo (preformación) y, por otro lado, esa información genética controla espacial y temporalmente los procesos que constituyen las sucesivas etapas del desarrollo como liberación de las potencialidades contenidas en la célula inicial única (epigénesis) (62).
En Embriología, por “epigénesis” – en contraposición a la “preformación”– se entiende la teoría de que las estructuras nuevas y los organismos se desarrollan a partir de una masa indiferenciada original de materia viva en el curso del desarrollo embrionario. Waddington (63) definió ya en los años 1939 y 1940 la “epigenética” como la rama de la Biología que se ocupa del análisis causal del desarrollo, definiendo a su vez el “epigenotipo” como el sistema de desarrollo total que está compuesto por series de desarrollo interrelacionadas a través de las cuales se realiza la forma adulta de un organismo y que comprende la totalidad de las interacciones entre los genes y entre los genes y el ambiente no genético que da como resultado el fenotipo (“epifenotipo”). El epigenotipo de una célula es un carácter estable y heredable, al menos durante muchas generaciones celulares, cuyo modo de impresión está por encima o además del genotipo clásico, esto es, la secuencia de bases del ADN (64).
En tiempos recientes ha surgido dentro de la Genética un nuevo concepto de “epigenética”(65) en relación con los mecanismos genéticos que influyen en el fenotipo sin alterar las secuencias del ADN, siendo la metilación del ADN (generalmente de las citosinas) uno de los mecanismos epigenéticos más importantes (patrón de metilación del ADN o metiloma).
Mecanismos epigenéticos son también las alteraciones de la estructura o remodelación de la cromatina por modificación de las histonas (metilación, acetilación, fosforilación) (66). La estructura local de la cromatina es un estado epigenético que puede cambiar de forma reversible por diversos tipos de mecanismos (remodelación de la cromatina). El hecho de que la actividad génica diferencial –a la que hacíamos referencia al definir el concepto genético de desarrollo– no implique cambios en la secuencia original del ADN explica por qué son posibles los mecanismos de reprogramación celular tales como la clonación por transferencia nuclear y la inducción de células troncales pluripotentes que han sido objeto del Premio Nobel en Fisiología o Medicina 2012.
En mi opinión, la confusión que en ocasiones se produce en el debate actual sobre lo que se denomina el “estatuto del embrión humano” es que se argumenta que el simple programa genético que contiene el cigoto producido por la fecundación de los gametos es necesario pero no suficiente para que se realice el proceso de desarrollo ya que son necesarios también los fenómenos epigenéticos como, por ejemplo, el silenciamiento de la expresión de los genes por la metilación de su ADN, pero manteniendo la secuencia original del ADN que constituye el programa genético del organismo reunido en el cigoto tras el proceso de fecundación de los gametos.
8. EPÍLOGO
Una vez más, puedo terminar mi intervención en la Sesión Científica que la Real Academia Nacional de Farmacia dedica a glosar los Premios Nobel concedidos en el año mostrando mi orgullo de pertenecer a un ámbito del conocimiento científico como es la Genética merecedora del más alto reconocimiento como son los premios Nobel.
Así pues, repitiendo y actualizando lo que he venido diciendo en otras ocasiones, hoy, en 2012, podemos estar orgullosos los amantes de la Genética porque ya son 40 las veces en que el galardón Nobel ha correspondido a 89 científicos del campo de la Genética o ciencias afines.
De los 40 premios considerados, 31 pertenecen al ámbito de la Fisiología o Medicina, 8 a la Química y 1 de la Paz y, a su vez, de los 89 científicos galardonados, 70 corresponden a premios de Fisiología o Medicina, 18 de Química y 1 de la Paz.
Finalmente, me gustaría destacar que en los doce años transcurridos del siglo XXI se ha premiado la investigación genética en doce ocasiones: 2001, 2002, 2004, dos en 2006, 2007, dos en 2008, dos en 2009, 2011 y en 2012. Sin duda, es una década prodigiosa para la Genética como una Alicia en el “País de las maravillas moleculares” que diría Lewis Carroll. En 1995 inicié con mi discurso de ingreso en esta Real Academia (67) la historia “nobelada” de la Genética que tuve la oportunidad de actualizar doce años después (68), en 2007, y que he puesto al día en 2012. No obstante, al paso que vamos, es posible que pronto se vuelva a quedar viejo.
Permítaseme finalizar esta intervención preguntándome en voz alta ¿para cuándo espera la Institución Nobel premiar la investigación genómica? En mi opinión, de los grandes temas actuales de investigación dentro de la Genética, la Genómica es el único que falta por recibir el galardón Nobel. En ocasiones anteriores me he atrevido a citar a científicos como Craig J. Venter y Francis Collins como merecedores del premio Nobel. Me pregunto si la controvertida personalidad del primero puede ser la causa de que se esté retrasando la decisión porque es evidente que no se puede premiar este campo de la investigación genética dejando fuera al Dr. Venter. El tema es de urgente solución porque, como decían Lander y Weinberg en un artículo publicado en la revista “Science” (68), la Genómica ocupa hoy día el centro de la Biología (“Genómica: viaje al centro de la Biología”, parafraseando el título de la conocida novela de Julio Verne). En la misma línea apuntan las palabras de Victor McKusick (69), uno de los grandes científicos en el campo de la Genética Humana, cuando decía:
“los laboratorios genómicos serán el lugar de formación de los científicos del futuro: nueva raza de científicos preparados para capitalizar tanto la revolución de la Genética Molecular como la revolución de la computación. Ellos serán los líderes de la Biología del siglo XXI”.
9. REFERENCIAS
1. Lacadena, J.R.; Genética (4ª edición, Capítulo XIX); AGESA; Madrid, 1988; p 931-1057.
2. Gurdon, J.B.; Melton, D.A. Nuclear reprogramming in cells. Science 322, 1811-1815 (2008).
3. Ford, M. Science magazine’s top 10 breakthroughs of the year, Science (2008).
4. Briggs, R.; King, T.J. Transplantation of living nuclei from blastula cells into enucleated frog eggs. Proc. Nat. Acad. Sci. 38, 455-463 (1952). Briggs, R.; King, T.J. Factors affecting the tranplantibility of nuclei of frog embryonic cells. J. exp. Zool. 122, 485-506 (1953). King, T.J.; Briggs, R. Changes in the nuclei of differentiating gastrula cells, as demonstrated by nuclear transplantation. Proc. Nat. Acad. Sci. 41, 321-325 (1955). Briggs, R.; King, T.J. Nuclear transplantation studies on the early gastrula (Rana pipiens). I. Nuclei of presumptive endoderm. Devel. Biol. 2, 252-270 (1960). Di Berardino, M.A.; King, J.T. Development and cellular differentiation of neural nuclear transplants of known karyotype. Devel. Biol. 15, 102-128 (1967).
5. Gurdon, J.B. Adult frogs derived from the nuclei of single somatic cells. Devel. Biol. 4, 256-273 (1962).
6. Elsdale, T.R.; Fischberg, M.; Smith, S. A mutation that reduces nucleolar number in Xenopus laevis. Exp. Cell Res., 14:642-643 (1958).
7. Kobel, H.R.; Brun, R.B.; Fischberg, M. Nuclear transplantation with melanophores, ciliated epidermal cells, and the established cell line A-8 in Xenopus laevis. J. Embryol. Exp. Morphol. 29, 539-547 (1973).
8. Di Berardino, M.A.; Hoffner, N.J. The current status of cloning and nuclear reprogramming in amphibian eggs. En Differentiation and Neoplasia (Results and Problems in Cell Differentiation, vol. 11); (edited by McKinell, R.G.; Di Berardino, M.A.; Blumenfeld, M.; Bergard, R.D., Eds.; Springer Verlag; Berlin, 1980; p 53-64.
9. Frisén, J.; Lendahl, U.; Perimann, T. Mature cells can be reprogrammed to become pluripotent. The 2012 Nobel Prize in Physiology or Medicine – Advanced Information. http://Nobelprize.org (2012)
10. Waddington, C.H. The strategy of the genes; a discussion of some aspects of theoretical biology; Allen & Unwin; Londres, 1957.
11. McGrath, J.; Solter, D. Inability of mouse blastomere nuclei transferred to enucleated zygotes to support development in vitro. Science 226, 1317-1318 (1984).
12. Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H.S. Viable offspring derived from fetal and adult mammalian cells. Nature 385, 810-813 (1997).
13. Lacadena, J.R. Genética y Sociedad. Real Acad. Nac. Farmacia; Madrid, 2011; p. 147. (El contenido de este apartado está necesariamente basado, aunque debidamente actualizado, en el texto publicado dentro del Discurso leído por el autor en la Sesión Inaugural del Curso de la Real Academia Nacional de Farmacia celebrada el 13 de enero de 2011).
14. Ver revisiones por Hochedlinger, K.; Plath, K. Epigenetic reprogramming and induced pluripotency. Development 136, 509-523 (2009). Izpisúa Belmonte, J.C.; Ellis, J.; Hochedlinger, K.; Yamanaka, S. Induced pluripotent stem cells and reprogramming: seeing the science through the hype. Nature Reviews Genetics 10, 878-883 (2009). Hochedlinger, K. El poder terapéutico de nuestras células. Investigación y Ciencia (julio 2010), 24-31 (2010).
15. Takahashi, K.; Mitsui, K.; Yamanaka, S. Role of Eras in promoting tumour-like properties in mouse embryonic stem cells. Nature 423, 541-545 (2003).
16. Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K. et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113, 631-642 (2003).
17. Chambers, I.; Colby, D.; Robertson, M.; Nichols, J.; Lee, S.; Tweedie, S. et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113, 643-655 (2003).
18. Tada, M.; Takahama, Y.; Abe, K.; Nakatsuji, N.; Tada, T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr. Biol. 11, 1553-1558 (2001).
19. Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663-676 (2006).
20. Yamanaka, S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell 1, 39-49 (2007).
21. Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachenko, R.; Hieu, J. TC.; Jaenisch, R.; Plath, K.; Hochedlinger, K. Directly reprogrammed fibroblasts show global epigenetic remodelling and widespread tissue contribution. Cell Stem Cell 1, 55-70 (2007). Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318-324 (2007).
22. Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 448, 313-317 (2007).
23. Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres acquire embryonic stem cells characteristics in induced pluripotent stem cells. Cell Stem Cell 4, 141-154 (2009).
24. Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanak, I.; Ichisaka, T.; Kawamura, I.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 474, 225-229 (2011).
25. Okita, K.; Nakawaga, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 322, 949-953 (2008).
26. Carey, B.W.; Markoulaki, S.; Hanna, J.; Saha, K.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Reprogramming of murine and human and somatic cells using a single polycistronic vector. Proc Nat Acad Sci. 106, 157-162 (2009).
27. Huangfu, D.; Osafune, K.; Maehr, R.; Go, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nature Biotechnology 26, 1269-1275 (2008).
28. Kim, J.B.; Sebastiano, V.; Wu, G.; Araúzo-Bravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den Boom, D.; Meyer, J.; Hübner, K.; Bernemann, C.; Ortmeier, C.; Zenke, M.; Fleischmann, B.K.; Zaehres, H.; Schöler, H.R. Oct4-induced pluripotency in adult neural stem cells. Cell 136, 411-419 (2009).
29. Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; Siuzdak, G.; Schöler, H.R.; Duan, L.; Ding, S. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell doi: 10.1016/j.stem.2009.04.005 (2009).
30. Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; jonsdottir, G.A.; Ruotti, V.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Induced pluripotent stem cell lines derived from human somatic cells. Science 318,1917-1920 (2007).
31. Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent cells from adult human fibroblasts by defined factors. Cell 131, 1-12 (2007).
32. Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; González, F.; Vassena, R.; Bilic, J.; Pekarik, V.; Tiscornia, G.; Edel, M.; Boué, S.; Izpisúa Belmonte, J.C. Efficient and rapid generation of induced pluripotent ítem cells from human keratinocytes. Nature Biotechnology 26, 1276-1278 (2008).
33. Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; Daley, G.Q.; Brack, A.S.; Collins, J.J.; Cowan, C.; Schlaeger, T.M.; Rossi, D.J. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 1-13 (2010).
34. Alipio, Z.; Liao, W.; Roemer, E.J.; Waner, M.; Fink, L.M.; Ward, D.C.; Ma, Y. Reversal of hyperglycemia in diabetic mouse models using induced-pluripotent stem (iPS)-derived pancreatic β-like cells. Proc. Nat. Acad. Sci., doi: 10.1073/pnas. 1007384107 (2010).
35. Xu, D.; Alipio, Z.; Fink, L.M.; Adcock, D.M.; Yang, J.; Ward, D.C.; Ma, Y. Phenotypic correction of murine hemophilia A using an iPS cell-based therapy. Proc Nat Acad Sci 106, 808-813 (2009).
36. Alipio, Z.; Liao, W.; Roemer, E.J.; Waner, M.; Fink, L.M.; Ward, D.C.; Ma, Y. Reversal of hyperglycemia in diabetic mouse models using induced-pluripotent stem (iPS)-derived pancreatic β-like cells. Proc. Nat. Acad. Sci. doi:10.1073/pnas. 1007384107 (2010).
37. Tsuji, O.; Miura, K.; Okada, Y.; Fujiyohi, K.; Mukaino, M.; Nagoshi, N.; Kitamura, K.; Kumagai, G.; Nishino, M.; Tomisato, S.; Higashi, H.; Nagai, T.; Katoh, H.; Kohda, K.; Matsuzaki, Y.; Yuzaki, M.; Ikeda, E.; Toyama, Y.; Nakamura, M.;Yamanaka, S; Okano, H. Therapeutic potential of appropriately evaluated safe-induced pluripotent stem cells for spinal cord injury. Proc. Nat. Acad. Sci. doi: 10-1073/pnas.0910106107 (2010).
38. Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; Wichterl, H.; Henderson, E-CE.; Eggan, K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218-1221 (2008).
39. Park, I.H. et al. Disease-specific induced pluripotent stem cells. Cell 134, 877-886 (2008).
40. Ebert, A.D.; Yu, J.; Rose Jr, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277-281 (2009).
41. Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W. ; Mu, Y.L. et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527-539 (2010).
42. Rashid, S.T.; Corbineau, S.; Hannan, N. ; Marciniak, S.J. ; Miranda, E. ; Alexander, G. et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Invest. 120, 3127-3136 (2010).
43. Liu, G-H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S-L.; panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; Thompson, J.; Boue, S.; Fung, H.L.; Sancho-Martínez, I.; Zhang, K.; Yates, J. III; Izpisua Belmonte, J.C. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature doi:10.1038/nature09879 (2011)
44. Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J. ; Kesavan, J.; Poppe, D. et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 480, 543-546 (2011).
45. IsraeL, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C. et al. (2012) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216-220 (2012).
46. The HD iPSC Consortium. Induced pluripotente stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11, 264-278 (2012).
47. Liu, G-H; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F. ; Ruiz, X.S. ; Zhang, W. ; Wagner, U.; Kim, A.; Ren, B.; Li, Y.; Goebl, A.; Rupa, J.K.; Soligalla, D.; Dubova, I.; Thomson, J.; Yates III, J.; rodriguez, G.; Sancho-Martínez, E.I.; Izpisua Belmonte, J.C. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature doi:10.1038/nature 11557 (2012).
48. Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sanger, S. et al. Modelling squizophrenia using human induced pluripotent stem cells. Nature 473, 221-225 (2011).
49. Kennedy, D. Editorial. Breakthrough of the year. Science 318:1833 (2007). Ford, M. Science magazine’s top 10 breakthroughs of the year. Science (2008).
50. Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154-156 (1981).
51. Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cells derived from human blastocystes. Science 282, 1145-1147 (1998).
52. Cibelli, J.B.; Kiessling, A.A.; Cunniff, K.; Richards, C.; Lanza, R.P.; West, M.D. Somatic cell nuclear transfer in humans: Pronuclear and early embryonic development. E-biomed: The Journal of Regenerative Medicine 2, 25-31 (2001). Cibelli, J.B.; Lanza, R.P.; West, M.D.; Ezzell, C. The first human cloned embryo. Scient. Am. 286, 42-49 (2002).
53. NIH. http://www.ClinicalTrials.gov (diciembre 2012)
54. Hadorn, E. Problems of determination and transdetermination. Brookhaven Symp. Biol. No. 18 (Genetic control of differentiation), 148-161 (1965).
55. Frisén, J.; Lendahl, U.; Perimann, T. Mature cells can be reprogrammed to become pluripotent. The 2012 Nobel Prize in Physiology or Medicine – Advanced Information. http://Nobelprize.org (2012)
56. Zhou, Q.; Brown, J.; Kanarek, A.; rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature 455, 627-632 (2008).
57. Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudohf, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature online doi: 10.1038/nature 08797 (2010). Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q. et al. Induction of human neuronal cells by defined transcription factors. Nature 476, 220-223 (2011).
58. Szabo, E.; Rampalli, S.;Risueño,R.M.; Schnerch, A.; Mitchell, R.; Fiebig-Comyn, A.; Levadoux-Martin, M.; Bhatia, M. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature on line doi:10.1038/nature 09591 (2010).
59. Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375-386 (2010).
60. Song, K.H.; Nam, Y.J.; Luo, X.; Qi, X.X.; Tan, W.; Huang, G.N. et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599-604 (2012).
61. Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L. et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593-598 (2012).
62. Lacadena, J.R. Genética (4ª edición, Capítulo XIX), AGESA; Madrid, 1988; p 931-1057.
63. Waddington, C.H. An introduction to modern Genetics, Allen & Unwin; Londres, 1939. Waddington, C.H. The genetic control of wing development in Drosophila. J. Genet. 41, 75 (1940).
64. Rieger, R.; Michaelis, A.; Green, M.M. Glossary of Genetics and Cytogenetics (4th edition); Springer Verlag; Berlin-Heidelber-Nueva York, 1976 (existe una traducción en español por M.J. Puertas, Editorial Alhambra, 1982).
65. Passarge, E. Color atlas of Genetics (3rd Edition, revised and updated); Thieme; Stuttgart, Nueva York, 2007.
66. Lacadena, J.R. Historia “nobelada” de la Genética: Concepto y Método; Discurso leído en la Sesión del 14 de diciembre de 1995 para su ingreso como Académico de Número de la Real Academia de Farmacia; Madrid, 1995; p 76.
67. Lacadena, J.R. Conmemorando los 100 años del término “Genética” (1905-2005): Una historia “nobelada” de la Genética, Conferencia Plenaria pronunciada en el Congreso de la Sociedad Española de Genética, Almería 2005. Universidad de León, Secretariado de Publicaciones; 2007; p 109.
68. Lander, E.S.; Weinberg, R.A. Genomics: journey to the center of Biology. Science 287, 1777-1782 (2000).
69. McKusick, V. The Human Genome Project: Plans, status and applications in Biology. En Gene mapping. Using law and ethics as guides; Annas, G.J.; Elias, S. Eds; Oxford University Press; Oxford, 1992; p 18-42.