REVISIÓN |
Bases moleculares de la esquizofrenia
 Cecilio Giménez
Cecilio Giménez
Centro de Biología Molecular Severo Ochoa. Universidad Autónoma de Madrid-Consejo Superior de Investigaciones Científicas. Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER). Centro de Investigación Biosanitaria IdiPAZ.
e-mail: cgimenez@cbm.uam.es
Recibido el 11 de noviembre de 2012
An. R. Acad. Farm. 78, 4, 2012, 425-445
RESUMEN
La esquizofrenia es una enfermedad compleja que afecta a alrededor del 1% de la población mundial y constituye una de las más importantes causas de discapacidad crónica. Aunque su etiología es desconocida, la enfermedad implica diversas anomalías neuromorfológicas y neuroquímicas y se acepta que factores genéticos, bien solos o potenciados por factores ambientales y epigenéticos juegan un papel importante en su patogénesis. Numerosos estudios realizados durante los últimos cuarenta años han relacionado a alteraciones en la neurotransmisión mediada por aminas biógenas, la neurotransmisión glutamatérgica y gabaérgica con la patología de las psicosis esquizofrénicas. Recientemente, a través del conocimiento de genes de susceptibilidad así como de proteínas implicadas en la patología de la enfermedad, están permitiendo un diagnóstico precoz de la misma y el desarrollo de una nueva generación de compuestos que puedan actuar como antipsicóticos de una forma más selectiva. |
Palabras clave: Esquizofrenia; Psicosis; Antipsicóticos.
ABSTRACT
Molecular bases of schizophrenia
Schizophrenia is a complex disorder that affects about 1% of the world population and is one of the most important causes of chronic disability. Although its etiology is unknown, the disease involves various morphologic and neurochemical abnormalities and it it’s accepted that genetic factors, either alone or enhanced by environmental and epigenetic factors play a role in its pathogenesis. Numerous studies over the last forty years have involved alterations in biogenic amines mediated neurotransmission, GABAergic and glutamatergic neurotransmission to the pathology of schizophrenic psychoses. Recently, the knowledge of susceptibility genes and proteins involved in the pathology of the disease, are allowing early diagnosis and the development of a new generation of compounds that can act as antipsychotics more selectively. |
Keywords: Schizophrenia; Psychosis; Antipsychotics.
1. Introducción
La esquizofrenia es un síndrome complejo que afecta a alrededor del 1% de la población mundial; constituye una de las más importantes causas de discapacidad crónica, tiene consecuencias devastadoras para las personas que la padecen y su entorno familiar, y es la séptima enfermedad en costos económicos de nuestra sociedad.
Actualmente, el tratamiento sintomático de la esquizofrenia tiene un éxito sólo parcial por lo que la necesidad de desarrollar nuevas vías terapéuticas basadas en un mayor conocimiento de la etiología y patogénesis de la misma es, desde todo punto de vista, imprescindible.
Sin embargo, hasta muy recientemente, el avance sobre el conocimiento de la enfermedad ha resultado extraordinariamente penoso y lento debido a una serie de factores como la heterogeneidad en los fenotipos esquizofrénicos y la ausencia de lesiones patológicas claras como las que han proporcionado vías de estudio en enfermedades tipo Alzheimer, Parkinson y otros procesos neurodegenerativos.
Por otra parte, la investigación sobre el mecanismo de acción de fármacos utilizados en el tratamiento de la esquizofrenia tampoco ha proporcionado un conocimiento claro sobre la patogénesis de la misma.
Por último, no existen modelos de la enfermedad fiables puesto que es imposible reproducir en animales las características esenciales de trastornos psíquicos típicos de seres humanos (estados de afectividad, comunicación y relaciones sociales). En este sentido, como aproximaciones posibles, se utilizan experimentalmente algunos modelos como ratones deficientes en una de las subunidades del receptor de glutamato NMDA, o deficientes en el transportador vesicular de glutamato VGLUT2 que reproducen en parte la bioquímica de la enfermedad (1-3).
Muy recientemente, se ha publicado un modelo celular que consiste en reprogramar fibroblastos de individuos adultos controles y esquizofrénicos obteniendo células troncales pluripotentes, que más tarde eran diferenciadas a células con fenotipo neuronal en las cuales, mediante arrays, se han estudiado variación en la expresión de genes, crecimiento celular y respuesta al tratamiento con antipsicóticos atípicos (4,5).
En general, la esquizofrenia se contempla ahora como una enfermedad compleja con factores múltiples que contribuyen a su patogénesis. Los factores desencadenantes de la misma pueden ser tan diversos como infecciones, complicaciones obstétricas, consumo de drogas, etc., aunque siempre con un fondo genético importante.
Es cierto que en la esquizofrenia no se han encontrado marcadores claros del tipo cuerpos de inclusión, neuritas distróficas, o gliosis reactivas presentes en muchas enfermedades neurodegenerativas (6). Sin embargo, de una forma más o menos consistente, recientemente se ha descrito la evidencia de sutiles anomalías en la citoarquitectura de la sustancia gris entorrinal y otras regiones corticales, así como la existencia de neuronas aberrantes en la sustancia blanca subyacente a regiones como la corteza prefrontal, corteza temporal y regiones del hipocampo, como veremos más adelante (7-9).
Por otra parte, estudios ultraestructurales e inmunohistoquímicos demuestran la existencia de una disminución en el volumen del neuropilo cortical sin una pérdida neuronal apreciable que sugieren déficits cualitativos y cuantitativos en los procesos neuronales y de conectividad sináptica (10, 11).
2. Factores genéticos implicados en esquizofrenia
La esquizofrenia es una enfermedad con un fuerte componente genético, en la que el factor de herencia se calcula entre el 80-85%. Estudios genéticos de ligamiento y asociación han implicado a varios loci del genoma que parecen tener relación con genes que confieren un alto riesgo de esquizofrenia (12-15) (Figura 1).
Por otra parte, estudios en arrays comparando perfiles de expresión genética en diferentes zonas del cerebro de individuos afectados y controles, muestran datos, no siempre consistentes, de variaciones en la expresión de genes implicados en la neurotransmisión gabaérgica y glutamatérgica, transmisión sináptica, el metabolismo cerebral, o genes relacionados con el proceso de mielinización.
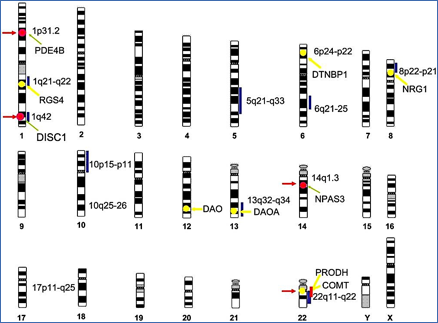
Figura 1.- Regiones cromosómicas y genes implicados en esquizofrenia. Líneas verticales azules: regiones cromosómicas con significancia en esquizofrenia. Líneas verticales rojas: deleciones cromosómicas. Flechas y círculos amarillos: genes identificados por estudios de ligamiento y asociación. Flechas y círculos rojos: genes identificados por estudio de traslocaciones cromosómicas. (De: Owen et al. 20).
A través de estos estudios genéticos, se están empezando a identificar proteínas codificadas por genes candidatos, como factores de riesgo en esquizofrenia tales como disbindina, neurorregulina 1, DAOA (factor activador de la D-amino oxidasa), COMT (catecol-O-metiltransferasa) y DISC (Disrupted in Schizophrenia). Entre todas ellas, parece que DISC, una proteína que se expresa en varias zonas del cerebro y que está involucrada en el crecimiento de neuritas y migración neuronal, se perfila como el mejor candidato para convertirse en un marcador fiable en la mayoría de los casos. A pesar de todo ello, la base genética de la enfermedad es compleja y la interpretación de los datos genéticos es difícil (16-20).
3. Características clínicas
Desde el punto de vista clínico, la esquizofrenia es un síndrome heterogéneo sin un síntoma o signo unitario definido y es imposible diagnosticarla a través de un test de diagnostico de laboratorio.
El diagnóstico, se realiza a través del estudio de una sintomatología individual que incluye la aparición de episodios temporales clasificados en diferentes categorías: síntomas positivos, que incluyen alucinaciones, ilusiones o paranoia; síntomas negativos, como el aislamiento social, la incapacidad para el afecto o la apatía; síntomas afectivos como depresiones o tendencia al suicidio y síntomas cognitivos, como alteraciones en la atención, la memoria y las funciones ejecutivas (Figura 2).
A pesar de todo ello, con frecuencia no resulta fácil diferenciarla de otros trastornos psicóticos como los bipolares.
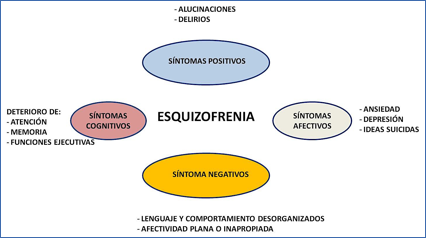
Figura 2.- Síntomas clínicos de la esquizofrenia.
La aparición de la esquizofrenia tiene lugar frecuentemente entre la segunda y la tercera década de la vida, aunque puede ocurrir en la niñez o en etapas adultas. Este hecho, unido a la aparición de signos tempranos, a veces muy sutiles, en capacidades cognitivas, de interacción social, funciones motoras y cambios morfológicos, que constituyen signos premonitorios de la enfermedad, sugiere que se trata de un problema de vulnerabilidad durante el desarrollo (21).
La adolescencia, es una etapa de la vida durante la cual se optimiza la red neuronal de forma que se mejoran la capacidad de juicio, las capacidades cognitivas y el control del comportamiento mediante el desarrollo de nuevos circuitos neuronales remodelando circuitos ya formados y descartando otros a través de un proceso de poda y maduración de los mismos.
4. Tratamiento farmacológico
Numerosos estudios realizados durante los últimos cuarenta años, relacionaron alteraciones de la neurotransmisión mediada por aminas biógenas con la patología de las psicosis esquizofrénicas.
De hecho, el primer tratamiento efectivo de la esquizofrenia, descubierto de manera fortuita a finales de los años cincuenta, fue demostrado mucho más tarde con la participación de receptores dopaminérgicos del tipo D2 en el mismo (22, 23).
La llamada “hipótesis dopaminérgica”, ha prevalecido hasta hace muy poco y se basaba en la observación de que los fármacos efectivos en el tratamiento de la enfermedad como el haloperidol, la clorpromazina y la perfenazina (los llamados antipsicóticos “típicos”), son antagonistas de receptores dopaminérgicos del tipo D2. Estos antipsicóticos de primera generación, aunque efectivos para algunos síntomas de la enfermedad, producían efectos neurológicos agudos y crónicos indeseados, como temblor, rigidez, distonía y disquinesia.
La segunda generación de antipsicóticos como la clozapina o la olanzapina (los llamados “atípicos”), reducen de forma considerable los efectos adversos antes citados y son más efectivos en el tratamiento de la esquizofrenia, posiblemente por su falta de especificidad para receptores dopaminérgicos D2, y su afinidad adicional para receptores serotonérgicos del tipo 5HT2A. De cualquier manera, el tratamiento con antipsicóticos atípicos conlleva un alto riesgo de desarrollar obesidad, hiperlipemia y diabetes tipo 2.
El conocimiento a nivel molecular del sitio de acción de los antipsicóticos, principalmente los receptores de dopamina, junto con la observación de que agonistas dopaminérgicos indirectos como la cocaína o las anfetaminas, así como alucinógenos de la familia del ácido lisérgico, que actúan sobre los transportadores neuronales de dopamina elevando el tono dopaminérgico, eran bien conocidos por inducir estados de psicosis en humanos (síntomas positivos clásicos de la esquizofrenia), apoyó la idea de que alteraciones en niveles de dopamina constituían el agente causal de la enfermedad (23).
5. Hipótesis dopaminérgica de la esquizofrenia
En un periodo de casi cuarenta años, numerosas compañías farmacéuticas han desarrollado compuestos que, actuando sobre receptores dopaminérgicos D2, sean efectivos como antipsicóticos. Sin embargo, todos estos fármacos presentan deficiencias en su acción en varios sentidos. Aproximadamente el 30% de los pacientes con esquizofrenia no experimentan mejoría alguna tras el tratamiento. Por otra parte, los antagonistas de los receptores D2 solamente son efectivos para el tratamiento de síntomas positivos de la enfermedad, mientras que los síntomas negativos y el déficit cognitivo permanecen. Más aún, tal como se indicaba más arriba, los efectos secundarios derivados del tratamiento son muy numerosos e incluyen sedación, ganancia de peso, disfunción sexual y toda una gama de síntomas propios de enfermedades como la diabetes, el Parkinson y la enfermedad de Alzheimer (para una revisión ver 24).
La razón de la cantidad de efectos colaterales de estos tratamientos se conoce ahora tras el conocimiento de la heterogeneidad de receptores dopaminérgicos existentes, D1-D5, todos metabotrópicos aunque con mecanismos de acción antagónicos, lo que unido a la falta de especificidad de los fármacos utilizados da como resultado la variedad de efectos indeseados de los mismos.
6. Hipótesis glutamatérgica de la esquizofrenia
Todos los datos acumulados durante años de práctica clínica en el tratamiento de la esquizofrenia, junto con datos experimentales más recientes, llevaron al convencimiento de que la hipótesis de la hiperfunción dopaminérgica constituía sólo una parte de la etiología de la enfermedad.
Datos más recientes obtenidos en estudios con animales de experimentación, han llevado a la idea generalizada de que una hipofunción en los receptores de glutamato del tipo NMDA está involucrada en la etiología de la esquizofrenia. Ratones con niveles de la subunidad NR1 de estos receptores ligeramente inferiores a los normales, y ratones carentes de la subunidad NR2 muestran un comportamiento muy similar a síntomas clínicos de la esquizofrenia que pueden ser atenuados mediante el tratamiento con antipsicóticos.
Por otra parte, animales genéticamente modificados en el sitio de unión de glicina en la subunidad R1 del receptor, muestran alteraciones en procesos de potenciación a largo plazo y de aprendizaje. Estos datos, primariamente observados en animales, han sido repetidamente comprobados en grupos de humanos clínicamente controlados o en individuos consumidores de fenilciclidina como droga de abuso, los cuales presentan síntomas clínicamente indistinguibles de individuos esquizofrénicos.
Por otro lado, y en sentido contrario, hay evidencias clínicas de que la administración a pacientes esquizofrénicos de co-agonistas de receptores NMDA les hacen mejorar, aunque de forma modesta, de muchos de los rasgos característicos de la enfermedad. Estos datos, junto con otros derivados de la clínica, son los que apoyan el papel de los receptores NMDA en esquizofrenia, y el convencimiento de que agentes capaces de potenciar la actividad de estos receptores pueden mejorar los síntomas positivos, negativos y cognitivos de la enfermedad (25-26).
Todo ello, ha llevado al convencimiento de que la neurotransmisión glutamatérgica está implicada en la esquizofrenia, en lo que actualmente se conoce como “hipótesis de la hipofunción de los receptores de NMDA”. Esta hipótesis se ha visto reforzada por el descubrimiento de varios genes de susceptibilidad relacionados con las vías glutamatérgicas como G72, NRG1, GRIA4, GRM3, GRM8, GRIN2D, o GRIN2A (27-32).
De cualquier forma, la hipótesis glutamatérgica y la dopaminérgica no son mutuamente excluyentes ya que, por ejemplo, la liberación de glutamato está regulada por receptores presinápticos de dopamina D2 en las vías corticolímbicas y corticoestriatales (33, 34). La interacción entre vías glutamatérgicas y dopaminérgicas tiene como consecuencia que la elevación de los niveles de glutamato y de su coagonista glicina, como veremos más adelante, puedan ser beneficiosos en el tratamiento de la enfermedad. De hecho, en este momento hay un número considerable de compuestos en fase clínica de investigación, que actúan sobre la neurotransmisión glutamatérgica, y más concretamente sobre el receptor metabotrópico GRM3 o sobre el sitio de unión de glicina al receptor de NMDA (35-38).
En la Figura 3 el esquema de un corte sagital de un cerebro de rata muestra la interacción entre vías dopaminérgicas, glutamatérgicas y gabaérgicas implicadas en psicosis.
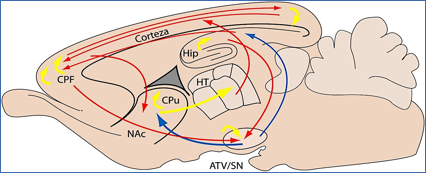
Figura 3.- Esquema de un corte sagital de cerebro de rata en el que se indican las principales vías de neurotransmisión involucradas en psicosis. Líneas rojas, glutamatérgicas; azules, dopaminérgicas; amarillas, gabaérgicas. ATV, área ventral tegmental; SN, substancia nigra; HT, hipotálamo; NAc, núcleo acumbens; Cpu, caudado putamen; CPF, corteza prefrontal; Hp, hipocampo.
Abundando en la idea de la interacción entre las hipótesis dopaminérgica y glutamatérgica de la enfermedad, hoy se sabe experimentalmente que una hipofunción en la actividad de receptores NMDA podría ser responsable de una situación hiperdopaminérgica. El flujo de dopamina aumenta tras el tratamiento con agentes antagonistas de NMDA y, por otra parte, agentes antipsicóticos atípicos revierten los efectos psicomiméticos producidos por antagonistas del receptor de glutamato. Estos efectos cruzados en sinapsis complejas en las que en una misma dendrita coinciden contactos de neuronas dopaminérgicas y glutamatérgicas dan como resultado cambios en la morfología de las neuronas y en la remodelación de circuitos (27).
En la Figura 4 se muestra la interacción entre receptores de glutamato NMDA y dopaminérgicos D1 y sus efectos a corto plazo a través de fosforilación directa de proteínas y a largo plazo mediante regulación de la síntesis de proteínas, cambiando la morfología de las neuronas.
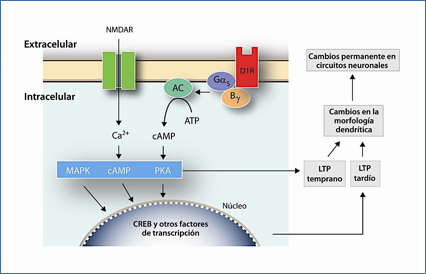
Figura 4.- Acciones sinérgicas de receptores de glutamato del tipo NMDA con receptores dopaminérgicos D1.
Lo más sorprendente desde el punto de vista celular es la demostración de que neuronas, en principio consideradas inequívocamente dopaminérgicas del mesencéfalo, se comportan como ambivalentes tanto dopaminérgicas como glutamatérgicas. Presentan un terminal típicamente glutamatérgico y unas extensiones laterales con varicosidades conteniendo estructuras pre y post sinápticas típicamente dopaminérgicas (39).
La Figura 5 resume de forma esquemática una visión de las interacciones en contactos de neuronas dopaminérgicas del área tegmental central que se proyectan hacia la corteza. Reciben contactos con neuronas gabaérgicas y glutamatérgicas e integran estas señales de una forma coherente. Por otra parte, cada una de esos tipos neuronales contienen específicamente receptores de una gran cantidad de tipos los cuales pueden unirse a sus sustratos y a otras sustancias con efectos sobre el sistema nervioso como etanol, nicotina, anfetaminas, cocaína, cannabis, etc.
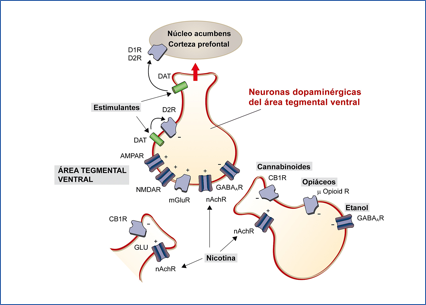
Figura 5.- Interacción de vías de neurotransmisión. Concurrencia de contactos sinápticos de vías glutamatérgicas y GABAérgicas con neuronas dopaminérgicas del área tegmental central.
Los receptores NMDA, puesto que poseen tantos y tan variados sitios moduladores de regulación deberían ofrecer, al menos en teoría, muchas posibilidades de manipulación farmacológica (Figura 6).
Constituyen unos detectores de excepción que requieren la concurrencia de tres factores para su activación: la unión de glutamato, la unión de glicina (o D-serina) y una despolarización previa a través de receptores AMPA. Sin embargo, todos los agonistas directos de los receptores sintetizados y ensayados hasta el momento han resultado ser neurotóxicos o conllevan efectos indeseados paralelos muy importantes (30).
Una vía indirecta de activación de los receptores NMDA parece ser más efectiva. Se trata de intervenir a través de sitios moduladores indirectos como el sitio de unión de aminas y, sobre todo, el sitio del co-agonista obligado glicina. A través de este mecanismo, los niveles de glutamato no se alteran o son regulados extracelularmente por sus transportadores. Los primeros ensayos clínicos administrando glicina a pacientes esquizofrénicos han dado resultados modestos, posiblemente debido a la baja penetración de la glicina, aunque indefectiblemente apoyan la hipótesis glutamatérgica de la enfermedad. Mucho más efectivos parecen ser las aproximaciones derivadas del aumento de niveles sinápticos de glicina mediante la inhibición de sus sistemas de recaptura (37, 38).
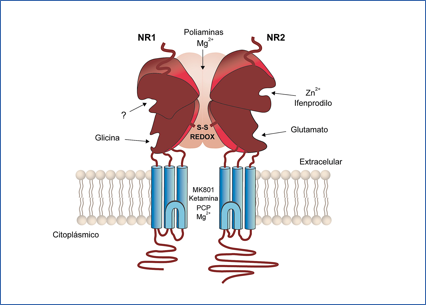
Figura 6.- Esquema de un receptor de glutamato ionotrópico del tipo NMDA. (De: Kemp, J.A. & McKernan, R.M. 2002, Nature Neuroscience 5, 1039-1042).
7. Bases celulares de la esquizofrenia: teoría gabaérgica
Mientras que las hipótesis dopaminérgica y glutamatérgica de la esquizofrenia han sido originadas partiendo de datos farmacológicos, la reciente implicación del GABA en la patogénesis de la enfermedad ha venido de la mano de datos neuropatológicos en pacientes durante la adolescencia.
Muy recientemente un grupo pionero en el campo se ha centrado en el estudio de una zona del cerebro, la corteza prefrontal dorsolateral (40). Se trata de una región formada por varias capas que es crucial para ir construyendo los cimientos de la experiencia, la memoria, el pensamiento y las emociones en una forma coherente, conformando al fin una visión consistente del mundo circundante. Esta zona del cerebro sufre una gran cantidad de cambios en sus circuitos durante la niñez y la adolescencia.
El grupo de Lewis se ha centrado en dos tipos celulares, las células piramidales de la corteza y las llamadas células en candelabro, unas interneuronas adyacentes a las anteriores en principio gabaérgicas (Figura 7).
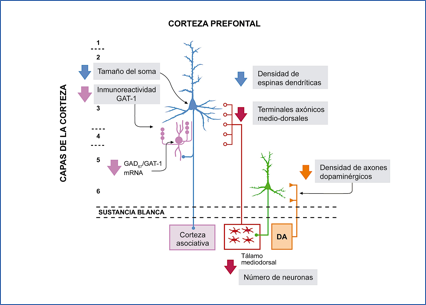
Figura 7.- Circuitos neuronales corticales en esquizofrenia. En azul, neuronas piramidales de la capa 3. En rosa, interneuronas en candelabro. Se esquematizan las interrupciones en conectividad entre los núcleos talámicos mediodorsales (MD) y las neuronas de la corteza prefrontal dorsolateral. (De: Ross et al. 6).
Numerosos estudios postmorten han mostrado que en esquizofrenia las células piramidales de esta zona tienen cuerpos más pequeños y un menor número de espinas dendríticas. Muchos investigadores sospechaban que este escaso número de ramificaciones era el resultado de un proceso aberrante de eliminación de circuitos durante la adolescencia. El proceso de poda o eliminación de sinapsis tiene por objeto eliminar las más débiles y dejar las más potentes. La sospecha era que en esquizofrenia este proceso no discrimina y elimina tanto sinapsis débiles como potentes (41-44).
El mismo grupo demostró recientemente que esto no es cierto del todo. La mayor parte de las sinapsis de la capa 3 del área en cuestión están ya maduras funcionalmente antes del proceso de poda. La hipótesis de este grupo es que las sinapsis de la capa 3 ya funcionales son más débiles en esquizofrénicos antes de comenzar la eliminación de sinapsis, por ello cuando empiezan a desaparecer sinapsis durante la poda, se ponen de manifiesto los problemas clínicos porque no hay sinapsis de reserva para equilibrar la pérdida (45-48).
En las células en candelabro se ha detectado una pérdida de la cantidad de transportadores de GABA, posiblemente relacionada con una disminución en la señalización por BDNF o una hipofunción de los receptores del tipo NMDA. Consistente con esta disminución en la neurotransmisión gabaérgica, se ha encontrado una up-regulación de receptores postsinápticos del tipo GABA-A.
Esto afecta al desarrollo de las células piramidales y, a la postre, a los contactos con la corteza asociativa y al número de neuronas del tálamo mediodorsal (49-55) (Figura 7).
La hipótesis de Lewis es que durante la niñez o al principio de la adolescencia las células en candelabro fallan de alguna manera en sus contactos con las piramidales y esto interfiere en la construcción de contactos y redes necesarios para hacer sinapsis robustas. Todo ello contribuye a que en la zona afectada se produzcan redes incapaces de generar contactos coordinados y vigorosos que generen la llamada memoria de trabajo. El resultado, sutil pero aparente, es que la pérdida de sinapsis que son podadas durante la adolescencia acaba generando un cerebro incapaz de organizar eléctricamente los pensamientos de una forma lógica.
8. Dianas terapéuticas basadas en modelos glutamatérgicos
En todo este complejo panorama, aparecen como claros signos implicados en esquizofrenia alteraciones funcionales en la neurotransmisión dopaminérgica, gabaérgica y glutamatérgica, aparte de sutiles cambios morfológicos en etapas tempranas. Nuestro grupo se ha centrado en el estudio de la neurotransmisión glutamatérgica y glicinérgica principalmente.
Desde hace años hemos estudiado dos proteínas que de forma diferente pero complementaria regulan la actividad de sinapsis glutamatérgicas: el transportador de glutamato GLT1 y el transportador de glicina GLYT1.
La Figura 8 muestra un esquema simple de la organización de una sinapsis glutamatérgica típica. En este esquema aparecen las dos proteínas a las que antes aludía. Un transportador de glutamato implicado directamente en enfermedades como la esquizofrenia o la esclerosis lateral amiotrófica (56), y un transportador de glicina GLYT1 que regula su concentración en las inmediaciones de los receptores NMDA (30, 57-59).
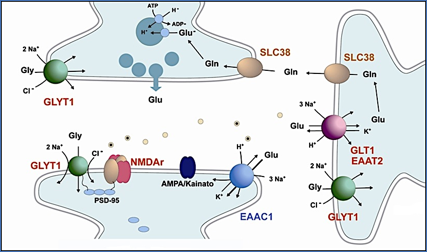
Figura 8.- Esquema de una sinapsis glutamatérgica. Están representadas algunas de las proteínas típicas de este tipo de sinapsis. GLYT1, transportador de glicina. GLT1, transportador de glutamato. Receptores e glutamato del tipo AMPA y NMDA.
Nuestro grupo, junto con un grupo noruego, describió hace años la distribución regional y celular de los transportadores de glicina (60, 61). Concretamente, la proteína GLYT1 parecía ser exclusivamente de origen glial frente a nuestro anticuerpo generado contra el extremo carboxilo de la proteína. De cualquier manera, detectamos mensajeros de la misma en células neuronales. Esta contradicción se aclaró diez años después tras generar un nuevo anticuerpo contra el extremo amino de la misma.
A través de experimentos de inmunohistoquímica identificamos y localizamos al transportador de glicina GLYT1 en elementos neuronales del cerebro estrechamente relacionados a vías glutamatérgicas (62). Por otra parte, mediante microscopía electrónica pudimos hacer una localización ultraestructural de GLYT1. La tinción aparecía en terminales sinápticos de sinapsis asimétricas de muchos elementos neuronales. Con técnicas de microscopía electrónica post-embeding pudimos ver la acumulación de partículas de oro en densidades postsinápticas y en zonas activas presinápticas.
Por otra parte, mediante estudios bioquímicos y de biología molecular y celular demostramos la colocalización de GLYT1 y GLT1 con la proteína de andamiaje PSD95 y a su vez con los receptores NMDA (63, 64). Experimentos de microscopía confocal utilizando diferentes fluoróforos demostraron claramente la interacción de GLT1 con PSD95 en espinas dendríticas. En estos trabajos no sólo vimos la co-localización de estas proteínas con técnicas de biología celular, sino que demostramos los determinantes estructurales en cada una de ellas necesarios para la interacción proteína-proteína (63, 64).
En el esquema de la Figura 9, que resume resultados de varios años, muestra cómo el transportador de glicina GLYT1 y el receptor NMDA se encuentran en neuronas asociados a través de la proteína de andamiaje PSD95 en espinas dendríticas. Todo apuntaba a que estas proteínas físicamente en contacto deberían funcionar de una forma coordinada.
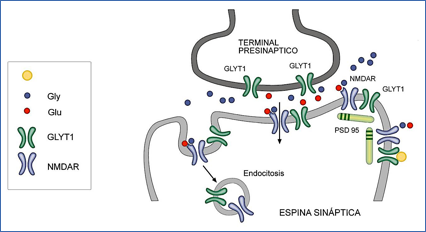
Figura 9.- Colocalización del transportador de glicina GLYT1 con el receptor NMDA y con la proteína de andamiaje PSD95. El esquema representa un terminal glutamatérgico de una espina sináptica con las proteínas GLYT1 y NMDA unidas a PSD95.
9. Inhibidores del transportador de glicina GLYT1
La activación de receptores NMDA requiere su unión simultánea a los neurotransmisores glutamato y glicina. Datos sobre la manipulación de la actividad de los transportadores de glutamato para aumentar la actividad de los receptores NMDA han resultado fallidos porque incrementaban la neurotoxicidad ejercida por el glutamato de forma similar a como la hacían los compuestos bloqueantes del canal (26, 65-67). Desde hace algunos años se pensó que la inhibición farmacológica del transportador de glicina GLYT1 aumentaría la concentración de la misma en sinapsis glutamatérgicas y, así, la actividad de los receptores NMDA. Por ello todos los esfuerzos se han centrado en la regulación indirecta de los receptores NMDA a través del transportador de glicina GLYT1.
Numerosos laboratorios farmacéuticos han desarrollado en los últimos años sustancias capaces de inhibir la actividad de GLYT1, muchas de las cuales están en fases clínicas avanzadas próximas a salir al mercado como una nueva generación de antipsicóticos, administrados solos o junto a reguladores del receptor metabotrópico de glutamato mGluR5 o junto a antipsicóticos atípicos (67, 68-72). En términos generales, se han tomado tres sustancias como base para la síntesis de inhibidores de GLYT1: la glicina, la sarcosina (N-metilglicina) y la glicildodecilamida.
10. Moduladores alostéricos positivos de receptores glutamatérgicos mGluR5
Actualmente, una segunda alternativa para el desarrollo de nuevas sustancias capaces de actuar en sinapsis glutamatérgicas es a través de la activación de receptores acoplados a proteína G, como los receptores metabotrópicos de glutamato pre y postsinápticos. La unión de glutamato a sus receptores modula la liberación del mismo a la sinapsis y/o la respuesta postsináptica al mismo, regulando así la fuerza de la señal. Todos estos receptores pertenecen a la familia de proteínas de siete dominios transmembrana con el extremo amino localizado extracelularmente formando parte de un gran dominio de unión al sustrato y el extremo carboxilo en el interior celular. Se han clonado siete receptores metabotrópicos de glutamato que clasifican en tres grupos dependiendo de su similitud estructural, propiedades farmacológicas y mecanismo de transducción de la señal.
Numerosos estudios han demostrado que la activación del receptor mGluR5, perteneciente al grupo I y con localización postsináptica, potencia la acción de receptores NMDA. Esta acción parece ser específica, puesto que la activación de receptores de los grupos II y III no varía las corrientes mediadas por NMDA, y la activación de receptores del grupo I no lo hacen con corrientes mediadas por receptores tipo AMPA. Todo ello apoya la idea de que una activación selectiva de receptores mGluR5 podría ser capaz de normalizar la actividad de receptores de glutamato NMDA funcionalmente hipofuncionales y constituir un nuevo grupo de sustancias con utilidad terapéutica en tratamientos antipsicóticos.
Hasta el momento se han desarrollado varias estrategias para la síntesis de moduladores alostéricos positivos de estos receptores con resultados aceptables. Una ventaja funcional que presentan estos compuestos es que al actuar alostéricamente, no en el sitio de unión del sustrato, requieren la presencia del ligando agonista endógeno. Puesto que los neurotransmisores son liberados a pulsos y rápidamente retirados del medio, la presencia de moduladores alostéricos que actúan únicamente cambiando la sensibilidad del receptor por su agonista, permitiría al sistema responder de una forma “más fisiológica” regulando las concentraciones de glutamato en el medio (para una revisión ver 28).
11. Conclusiones y perspectivas de futuro
La esquizofrenia es una enfermedad compleja y discapacitante con una prevalencia mayor que enfermedades como la diabetes tipo 1 o la enfermedad de Alzheimer. Las terapias paliativas que se están utilizando en la actualidad, desarrolladas a partir de la llamada “hipótesis dopaminérgica”, son efectivas paliando solo los llamados síntomas negativos de la enfermedad y conllevan efectos secundarios indeseados importantes.
El conocimiento en los últimos años de genes de susceptibilidad y de proteínas implicadas en la esquizofrenia está permitiendo el desarrollo de nuevas vías de actuación terapéutica. En este campo la industria farmacéutica ha hecho avances importantes con el desarrollo de sustancias basadas en la “teoría glutamatérgica de la esquizofrenia” que actúan modulando la función de receptores tipo NMDA.
La potenciación de este tipo de receptores se puede conseguir por dos vías: incrementando los niveles de glicina en su entorno en el espacio intersináptico mediante la inhibición del transportador de glicina del tipo GLYT1, o bien mediante la activación de receptores postsinápticos de glutamato mGluR5 a través de una modulación alostérica positiva. Compuestos de estas dos familias se encuentran actualmente en fases clínicas de validación.
Por último, hay que resaltar el esfuerzo enorme de muchos grupos de investigación en los últimos años durante los cuales se han hecho avances importantes en el análisis de fenotipos, análisis por neuroimagen, genética y conocimiento de la patología molecular de la esquizofrenia, lo que hace que el problema de esta enfermedad se vea actualmente con un cierto optimismo.
En términos generales se puede decir que la esquizofrenia se contempla ahora no como un desorden genético estático, sino como un proceso dinámico y sutil del desarrollo del cerebro en cuya etiología están presentes alteraciones en conjuntos de genes, bien por mutaciones o polimorfismos, así como factores epigenéticos que determinan una vulnerabilidad genética a la enfermedad.
12. REFERENCIAS
1. Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.; Bristol, L. A.; Jin, L.; Kuncl, R. W.; Kanai, Y.; Hediger, M. A.; Wang, Y.; Schielke, J. P. & Welty, D. F. (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate, Neuron 16, 675-686.
2. Belforte, J. E.; Zsiros, V.; Sklar, E. R.; Jiang, Z.; Yu, G.; Li, Y.; Quinlan, E, M. & Nakazawa, K. (2010) Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes Nature Neuroscience 13, 76-83.
3. Wallén-Mackenzie, A.; Wootz, H. & Englund, H. (2010) Genetic inactivation of the vesicular glutamate transporter 2 (VGLUT2) in the mouse: what have we learnt about functional glutamatergic neurotransmission? Ups Journal of Medical Sciences 115, 11-20.
4. Brennand, K. .; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; McCarthy, S.; Sebat, J. & Gage, F. H. (2011) Modelling schizophrenia using human induced pluripotent stem cells. Nature 12, 473, 221-225. Erratum in: Nature. 2011, 24; 479, 556.
5. Brennand, K.J. & Gage, F. H. (2012) Modeling psychiatric disorders through reprogramming Diseases Models Mechanisms 5, 26-32.
6. Ross, D. E. & Margolis. R. L. (2005) Neurogenetics: insights into neurodegenerative diseases and approaches to schizophrenia. Clinical Neuroscience Research 5, 3-14.
7. Antonova, E.; Sharma, T.; Morris, R. & Kumari, V. (2004) The relationship between brain structure and neurocognition in schizophrenia: a selective review. Schizophrenia Research 70, 117-145.
8. Honea R, Crow TJ, Passingham D, Mackay CE. (2005) Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. American Journal of Psychiatry 12, 2233-2245.
9. Dickey, C.C.; McCarley, R.W.; Voglmaier, M.M.; Niznikiewicz, M. A.; Seidman, L. J.; Demeo, S.; Frumin, M.& Shenton, M.E. (2003) An MRI study of superior temporal gyrus volume in women with schizotypal personality disorder. American Journal of Psychiatry 160, 2198-2201.
10. Selemon, L.D & Goldman-Rakic, P.S. (1999) The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biological Psychiatry 45, 17-25.
11. Selemon, L. D.; Rajkowska, G. & Goldman-Rakic, P. S. (1998) Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of three-dimensional, stereologic counting method. The Journal of Comparative Neurology. 392, 402-412.
12. Chen, J.; Lipska, B. K. & Weinberger, D. R. (2006) Genetic mouse models of schizophrenia: from hypothesis-based to susceptibility gene-based models. Biological Psychiatry 59, 1180-1188.
13. Ripke, S.; Sanders, A. R.; Kendler, K. S.; et al. (2011) Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium.Genome-wide association study identifies five new schizophrenia loci. Nature Genetics 43, 969-976.
14. Craddock, N.; O'Donovan, M. C. & Owen, M. J. (2007) Phenotypic and genetic complexity of psychosis. Invited commentary on Schizophrenia: a common disease caused by multiple rare alleles. British Journal of Psychiatry 190, 200-203. Erratum in: Br J Psychiatry. 2007 190:365.
15. Hamshere, M. L.; Holmans, P. A.; McCarthy, G, M.; Jones, L. A.; Murphy, K, C.; Sanders, R. D.; Gray, M. Y.; Zammit, S.; Williams, N. M.; Norton, N.; Williams, H. J.; McGuffin, P.; O'Donovan, M. C.; Craddock, N.; Owen, M. J. & Cardno, A. G. (2011) Phenotype evaluation and genomewide linkage study of clinical variables in schizophrenia. American Journal of Medicine Genetic B Neuropsychiatry Genetic 156B, 929-940.
16. Owen, M.J. (2012) Implications of genetic findings for understanding schizophrenia. Schizophrenia Bulletin 38, 904-907.
17. Levinson, D.F.M.; Shi, J.; Wang. K.; Oh, S.; Riley, B. et al. (2912) The Schizophrenia Psychiatric GWAS Consortium. Genome-Wide Association Study of Multiplex Schizophrenia Pedigrees. American Journal of Psychiatry 169, 963-973.
18. Jia, P.; Wang, L.; Fanous, A. H.; Pato, C. N.; Edwards, T. L. et al (2012) International Schizophrenia Consortium, Zhao, Z. Network-assisted investigation of combined causal signals from genome-wide association studies in schizophrenia. PLoS Computational Biology 8, e1002587.
19. Doherty, J.L.; O'Donovan, M. C. & Owen, M. J. (2012) Recent genomic advances in schizophrenia Clinical Genetics 81, 103-109.
20. Owen, M.J.; Craddock, N. & O'Donovan, M. C. (2005) Schizophrenia: genes at last? Trends in Genetics 21, 518-525.
21. Niemi, L. T.; Suvisaari, J. M.; Tuulio-Henriksson, A. & Lönnqvist, J. K. (2003) Childhood developmental abnormalities in schizophrenia: evidence from high-risk studies. Schizophrenia Research 60, 239-258.
22. Snyder, S. H. (2006) Dopamine receptor excess and mouse madness. Neuron 49, 484-485.
23. Carlsson, A. (1988) The current status of the dopamine hypothesis of schizophrenia. Neurospsychopharmacology. 1, 179-186.
24. Javitt, D. C. (1987) Negative schizophrenic syntomatology and the PCP (phencyclidine) model of schizophrenia. Hillside Journal of Clinical Psychiatry 9, 12-35.
25. Lang, U. E.; Puls, I.; Müller, D. J.; Strutz-Seebohm, N. & Galliant, J. (2007) Molecular mechanisms of schizophrenia. Cellular Physiology and Biochemistry 20, 687-702).
26. Javitt, D. C. & Zukin, S. R. (1991) Recent advances in the phencyclidine model of schizophrenia. American Journal of Psychiatry 148, 1301-1308.
27. Javitt, D.C. (2007) Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. International Review of Neurobiology 78, 69-108.
28. Lindsley C. W.; Shipe W. D.; Wolkenberg S. E.; Theberge C. R.; Williams, D. L.; Sur, C. & Kinney G. G. (2006) Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Current Topics in Medical Chemistry 6, 771-785.
29. Watis, L.; Chen S. H.; Chua, H. C.; Chong, S. A. & Sim, K. (2008) Glutamatergic abnormalities of the thalamus in schizophrenia: a systematic review Journal of Neural Transmission 115, 493-511.
30. Millan, M. J. (2005) N-methyl-D-aspartate receptors as a target for improved antipsychotic agents: novel insights and clinical perspectives. Psychopharmacology 179, 30-53.
31. Olney, J. W.; Newcomer, J. W. & Farber, N. B. (1999): NMDA receptor hypofunction model of schizophrenia. Journal of Psychiatry Research 33, 523-533.
32. Moghaddam, B. (2003) Bringing order to the glutamate chaos in schizophrenia. Neuron 40, 881-884.
33. Goldstein, M. & Deutch, A. Y. (1992) Dopaminergic mechanisms in the pathogenesis of schizophrenia. FASEB Journal 6, 2413-2421.
34. Javitt, D. C.; Sershen, H.; Hashim, A. & Lajtha, A. (2000) Inhibition of striatal dopamine release by glycine and glycyldodecylamide. Brain Research Bulletin 52, 213-216.
35. Javitt, D. C. (2004) Glutamate as a therapeutic target in psychiatric disorders Molecular Psychiatry 9, 984-997.
36. Lechner, S. M. (2006) Glutamate-based therapeutic approaches: inhibitors of glycine transport. Current Opinion on Pharmacology. 6, 75-81.
37. Thomsen, C. (2006) Glycine transporter inhibitors as novel antipsychotics. Drug Discovery: Therapeutic Strategies 3, 539-545.
38. Lindsley, C. W.; Shipe, W. D.; Wolkenberg, S. E.; Theberge, C.R.; Williams, D. L. Jr.; Sur. C. & Kinney, G. G. (2006) Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Current Topics in Medicinal Chemistry 6, 771-785.
39. Descarries, L.; Bérubé-Carrière, N.; Riad, M.; Bo, G. D.; Méndez, J. A. & Trudeau, L. E. (2007) Glutamate in dopamine neurons: synaptic versus diffuse transmission. Brain Research Reviews 58, 290-302.
40. Millan, M. J. (2005) N-Methyl-D-aspartate receptors as a target for improved antipsychotic agents: novel insights and clinical perspectives. Psychopharmacology (Berlin) 179, 30-53.
41. Dobbs, D. (2010) Schizophrenia appears during adolescence. But where does one begin and the other end? Nature 468, 154-156.
42. Beneyto, M. & Lewis DA. (2011) Insights into the neurodevelopmental origin of schizophrenia from postmortem studies of prefrontal cortical circuitry. International Journal of Developmental Neuroscience 29, 295-304.
43. Sweet, R. A.; Fish, K. N. & Lewis, D. A. (2010) Mapping Synaptic Pathology within Cerebral Cortical Circuits in Subjects with Schizophrenia. Frontiers in Human Neuroscience 23, 44- 52.
44. Volk, D. W. & Lewis, D. A. (2010) Prefrontal cortical circuits in schizophrenia. Current Topics in Behavioral Neuroscience 4, 485-508.
45. Lewis, D. A. (2011) The chandelier neuron in schizophrenia. Developmental Neurobiology 71, 118-127.
46. Piper, M.; Beneyto, M.; Burne, T. H.; Eyles, D. W.; Lewis, D. A. & McGrath, J. J. (2012) The neurodevelopmental hypothesis of schizophrenia: convergent clues from epidemiology and neuropathology. Psychiatric Clinical North America 35, 571-584.
47. Lewis, D. A. (2012) Cortical circuit dysfunction and cognitive deficits in schizophrenia: implications for preemptive interventions. European Journal of Neuroscience 35, 1871-1878.
48. Glausier, J.R. & Lewis, D. A. (2012) Dendritic spine pathology in schizophrenia. Neuroscience. [Epub ahead of print] PubMed PMID: 22546337.
49. Gonzalez-Burgos, G. & Lewis, D. A. (2012) NMDA Receptor Hypofunction, Parvalbumin-Positive Neurons, and Cortical Gamma Oscillations in Schizophrenia. Schizophrenia Bulletin 38, 950-957.
50. Stan, A. D. & Lewis D. A. (2012) Altered cortical GABA neurotransmission in schizophrenia: Insights into novel therapeutic strategies. Current Pharmological Biotechnology 13, 1557-1562.
51. Curley, A. A. & Lewis, D.A. (2012) Cortical basket cell dysfunction in schizophrenia. Journal of Physiology 590 (Pt 4), 715-724.
52. Lewis, D. A.; Curley, A. A.; Glausier, J. R. & Volk, D. W. (2012) Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends in Neuroscience 35, 57-67.
53. Volk, D. W.; Eggan, S. M. & Lewis, D.A. (2010) Alterations in metabotropic glutamate receptor1α and regulator of G protein signaling 4 in the prefrontal cortex in schizophrenia. American Journal of Psychiatry 167, 1489-1498.
54. Gonzalez-Burgos, G.; Hashimoto, T. & Lewis, D. A. (2010) Alterations of cortical GABA neurons and network oscillations in schizophrenia. Current Psychiatry Reports 12, 335-344.
55. Lewis, D.A. (2009) Neuroplasticity of excitatory and inhibitory cortical circuits in schizophrenia. Dialogues on Clinical Neuroscience 11, 269-80.
56. Bristol, L. A. & Rothstein, J. D. (1996) Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Annals of Neurology 39, 676-679.
57. Zafra, F. & Giménez, C. (2008) Glycine transporters and synaptic function. IUBMB Life 60, 810-817.
58. Aragón, C. & López-Corcuera, B. (2005) Glycine transporters: crucial roles of pharmacological interest revealed by gene deletion. Trends in Pharmacological Sciences 26, 283-286.
59. Aragón, C. & López-Corcuera, B. (2003) Structure, function and regulation of glycine neurotransporters. European Journal of Pharmacology 31, 249-262.
60. Zafra, F.; Aragón, C.; Olivares, L.; Danbolt, N.C.; Giménez, C. & Storm-Mathisen, J. (1995) Glycine transporters are differentially expressed among CNS cells. Journal of Neuroscience 15, 3952-3969.
61. Zafra, F.; Gomeza, J.; Olivares, L.; Aragón, C. & Giménez, C. (1995) Regional distribution and developmental variation of the glycine transporters GLYT1 and GLYT2 in the rat CNS. European Journal of Neuroscience 7, 1342-1352.
62. Cubelos, B.; Giménez, C. & Zafra, F. (2005) Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cerebral Cortex 15, 448-459.
63. Cubelos, B.; González-González, I.M.; Giménez, C. & Zafra, F. (2005) The scaffolding protein PSD-95 interacts with the glycine transporter GLYT1 and impairs its internalization. Journal of Neurochemistry 95, 1047-1058.
64. González-González, I. M.; García-Tardón, N.; Cubelos, B.; Giménez, C. & Zafra, F. (2008) The glutamate transporter GLT1b interacts with the scaffold protein PSD-95. Journal of Neurochemistry 105, 1834-1848.
65. Krystal, J.H.; Anand, A. & Moghaddam, B. (2002) Effects of NMDA receptor antagonists:implications for the pathophysiology of schizophrenia. Archives of General Psychiatry 59, 663-664.
66. Abi-Saab, W.M.; D'Souza, D. C.; Moghaddam, B. & Krystal, J. H. (1998) The NMDA antagonist model for schizophrenia: promise and pitfalls. Pharmacopsychiatry 31 Suppl2,104-109.
67. Krystal, J.H.; Karper, L. P.; Seibyl, J. P.; Freeman, G. K.; Delaney, R.; Bremner, J. D.; Heninger, G. R.; Bowers, M. B. Jr. & Charney, D. S. (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Archives of General Psychiatry 51, 199-214.
68. Sur, C. & Kinney, G. G. (2007) Glycine transporter 1 inhibitors and modulation of NMDA receptor-mediated excitatory neurotransmission. Current Drug Target 8, 643-649.
69. Lindsley, C. W.; Zhao, Z.; Leister, W. H.; O'Brien, J.; Lemaire, W.; Williams. D. L. Jr.; Chen, T. B.; Chang, R, S.; Burno, M.; Jacobson, M. A.; Sur, C.; Kinney, G. G.; Pettibone, D. J.; Tiller, P. R.; Smith, S.; Tsou, N. N.; Duggan, M. E., Conn, P. J. & Hartman, G. D. (2006) Design, synthesis, and in vivo efficacy of glycine transporter-1 (GlyT1) inhibitors derived from a series of[4-phenyl-1-(propylsulfonyl)piperidin-4-yl]methylbenzamides. Chemical Medicine 8, 807-811.
70. Rogers, B. N. & Schmidt, C. J. (2006) Novel approaches for the treatment of schizophrenia. Annual Reports in Medicinal Chemistry 41, 1-21.
71. Sur, C. & Kinney, G. (2007) Glycine transporter 1 inhibitors and modulation of NMDA receptor-mediated excitatory neurotransmission. Current Drug Targets 8, 643-649.
72. Dohi, T.; Morita, K.; Kitiyama, T.; Motoyama, N. & Morioka, N. (2009) Glycine transporter inhibitors as a novel drug discovery strategy for neuropathic pain. Pharmacology and Terapeuthics 123, 54-79.