REVISIÓN |
Presente y Futuro en el Descubrimiento de Fármacos para la Enfermedad de Chagas
Nuria E. Campillo, Pedro González-Naranjo, Juan Antonio Páez*
Instituto de Química Médica. CSIC. Juan de la Cierva 3, 28006-Madrid. España.
Recibido el 31 de octubre de 2011.
e-mail: jpaez@iqm.csic.es
RESUMEN
La enfermedad de Chagas, también conocida como Tripanosomiasis Americana es una enfermedad parasitaria causada por el Trypanosoma cruzi. Se estima que alrededor de 7,7 millones de personas se encuentran infectadas y padecen la enfermedad de Chagas. Esta enfermedad silenciosa que esta estrechamente relacionada con la pobreza, es transmitida a los humanos por unos insectos que se encuentran exclusivamente en el continente americano, principalmente en áreas rurales con muy deficientes condiciones de salubridad. Los fármacos existentes (nifurtimox y benznidazol), no siempre disponibles, constituyen un tratamiento paliativo, pero no curan la enfermedad y no son aceptables desde un punto de vista terapéutico debido a sus efectos secundarios indeseables y a su falta de eficacia. Por tanto, es necesario el desarrollo urgente de nuevos tratamientos y por tanto, sería muy conveniente la utilización del diseño racional en todas las etapas. El diseño de fármacos es una tarea compleja que requiere la colaboración interdisciplinar de muchos especialistas en diferentes campos de la ciencia. El presente trabajo describe de manera estructurada las diferentes estrategias que se han utilizado y las que se pueden utilizar en el futuro para el descubrimiento de nuevos fármacos para la enfermedad de Chagas. Se recogen las estrategias más clásicas como el diseño de análogos, el cribado sistemático o el basado en la información biológica y los métodos más novedosos basados en lo que se conoce como quimioinformática. |
Palabras clave: Enfermedad de Chagas; T. cruzi; QSAR; Quimioinformática; Cribado virtual; Redes neuronales; Anclaje.
ABSTRACT
The Present and Future of Drug Discovery for Chagas Disease
Chagas disease, also known as American
trypanosomiasis, is caused by infection with the Trypanosoma
cruzi. The Pan American Health Organization (PAHO)
estimates that 7.7 million persons currently have T. cruzi infection in the 21
endemic countries. This disease
can be transmitted to humans by insect vectors that are found only in the
American continent, mainly, in rural areas with unhealthy housing conditions
where poverty is a general concern. Nifurtimox and benznidazole are the only
drugs used against this disease, but sometimes they are not available. The
treatment of Chagas disease with nifurtimox or benznidazole is unsatisfactory
because of their limited efficacy on the prevalent chronic stage of the
disease and their toxic side effects. |
Keywords: Chagas Disease; T. cruzi; QSAR; Cheminformatics; Virtual screening; Neural network; Docking.
1. introducciÓn
La enfermedad de Chagas, también llamada Tripanosomiasis Americana es una enfermedad parasitaria causada por un protozoo flagelado llamado Trypanosoma cruzi. Este parásito es transmitido a los huéspedes vertebrados por un insecto hematófago, el Triatoma infestans, conocido como vinchuca o chinche. La enfermedad de Chagas también puede transmitirse a través de transfusiones de sangre, de madres a hijos durante el embarazo, o con menos frecuencia, a través de trasplantes de órganos o alimentos contaminados. El parásito se reproduce en los tejidos internos y causa problemas en el corazón, el esófago, el colon y el sistema nervioso (1).
Esta enfermedad es una endemia parasitaria que afecta a unos 7,7 millones de personas en Latinoamérica, extendiéndose desde el sur de los Estados Unidos hasta Argentina y Chile. No existe vacuna contra la enfermedad de Chagas y las personas afectadas pueden volverse a infectar después de recibir tratamiento farmacológico (2).
El hecho de que la enfermedad de Chagas esté íntimamente relacionada con la pobreza ha podido ser una de las razones que ha originado la falta de interés en el desarrollo de nuevos medicamentos eficaces. Esto explica que se sigan utilizando fármacos que fueron comercializados en los años 70, como el nifurtimox y el benznidazol que no están aprobados por la FDA (Federal Drug Administration). En la actualidad, esta enfermedad está clasificada por la Organización Mundial de la Salud como una enfermedad extremadamente olvidada (3).
El diseño de nuevos fármacos es una tarea compleja que requiere la colaboración interdisciplinar de muchos especialistas en diferentes campos de la ciencia. En el descubrimiento de nuevos fármacos hay que considerar, además de las propiedades farmacológicas, otros aspectos de gran relevancia, como es el de las propiedades ADMET (A: absorción, D: distribución, M: metabolismo, E: excreción y T: toxicidad). Por tanto, antes de tomar en consideración a un nuevo cabeza de serie (lead compound) de acuerdo a sus propiedades farmacológicas es necesario que se incluya un estudio más profundo de las propiedades ADMET del hit.
2. DiseÑo de FÁrmacos
El diseño de nuevos fármacos para la enfermedad de Chagas puede abordarse mediante dos estrategias generales. La primera consiste en el aislamiento y estudio de productos naturales procedentes de la medicina tradicional o de otras fuentes y la segunda, se refiere al desarrollo de nuevos compuestos de síntesis química o de procedencia biotecnológica (4, 5).
El uso de productos naturales como estrategia en la búsqueda de nuevos hits para la enfermedad de Chagas se ha realizado fundamentalmente en productos naturales de origen vegetal, aunque también se han aislado algunos compuestos a partir de hongos o de especie marinas. Existen numerosas revisiones recientes que recogen los trabajos realizados en este campo (6-9). Únicamente citar algunas de las familias estudiadas y de las que se han encontrado moléculas activas como las leguminosas o fabáceas (Cassia fistula, Harpalyce brasiliana, Pterodon pubescens (10), solanáceas (Acnist arborescens, Physalis angulata)boragináceas (Cordia globosa), clusiáceas (Mammea americana) (11) y diversas especies de la familia de las piperáceas (Piper glabratum, P. acutifolium (12); P. gaudichaudianum, P. aduncum (13); P. crassinervium (13, 14), Peperomia blanda (15).
A partir de estas especies naturales han sido aisladas y caracterizadas una gran variedad de estructuras químicas con propiedades anti-T. cruzi (8). De entre todas ellos podemos citar como más representativas a los flavonoides como chalconas, flavonas o isoflavonas; terpenoides como derivados de diterpenos (icetexano, labdano, azorellano), triterpenos pentacíclicos o lactonas sesquiterpénicas; lignanos; cumarinas; catequinas; xantonas; arilfloroglucinoles; antraquinonas y naftilisoquinolinas (6-8) (Figura 1). Finalmente, mencionar que también se han obtenido derivados anti-T. cruzi aislados a partir de esponjas marinas (Agelas sp.) (16) y hongos (Basidiomycota fungi) (17).
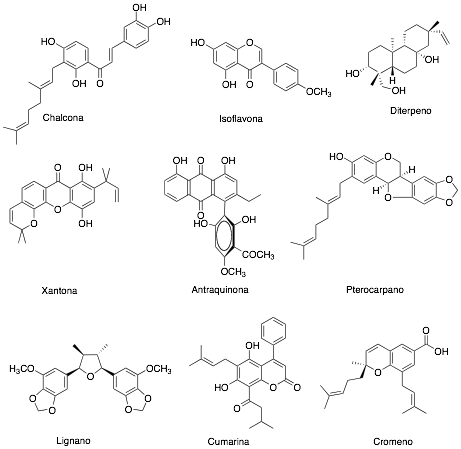
Figura 1.- Ejemplos representativos de productos naturales derivados de xantona (18), antraquinona (19), diterpeno (20), isoflavona (21) pterocarpano, chalcona (22), lignano (15), cumarina (11), cromeno (13).
Una estrategia más reciente utilizada en la búsqueda de nuevos hits es la que se refiere a la química combinatoria. Esta técnica fue desarrollada originalmente para la obtención de librerías con un número elevado de compuestos y para su posterior evaluación en diferentes dianas utilizando técnicas de HTS (high-throughput screening; acrónimo inglés de cribado de alto rendimiento). La aproximación inicial de la realización del cribado de alto rendimiento a miles de compuestos tiene bastantes detractores y la tendencia actual es la obtención de librerías más pequeñas de compuestos bien caracterizados.
Una clasificación más sistemática para el diseño de fármacos, basada en la metodología utilizada, podría establecerse en cuatro grandes apartados: diseño de análogos, diseño basado en cribado (screening) sistemático, diseño basado en información biológica y quimioinformática (Figura 2).
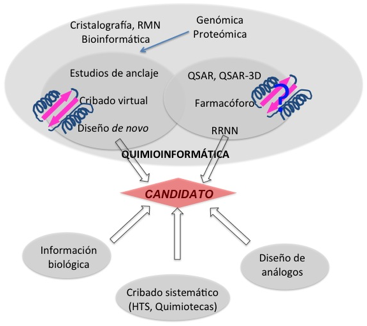
Figura 2.- Esquema general representativo en el diseño de fármacos.
2.1. Diseño de Análogos
Una de las estrategias más utilizadas para la obtención de derivados anti-T. cruzi ha sido el diseño de análogos de moléculas de probada actividad. Mediante esta estrategia moléculas con actividad biológica son modificadas para incrementar su potencia, su especificidad, mejorar su toxicidad, etc.
Desde el descubrimiento de la tripanosomiasis americana por Carlos Chagas en 1909 (23), un gran número de compuestos han sido sometidos a evaluación con el objetivo de encontrar moléculas que fueran activas frente al Tripanosoma cruzi. Entre las familias de derivados estudiadas se pueden citar derivados de fenotiazinas, acridinas, fenazinas, imidazoles, nitroimidazoles, triazoles, nitroimidazoles, tiadiazoles, isoxazoles, purinas, naftoquinonas, indoles, isoquinolinas, nitrofuranos, naftoquinonas, complejos metálicos etc. Las investigaciones realizadas utilizando métodos empíricos condujeron al descubrimiento de un nitrofurano (nifurtimox, Lampit) y un nitroimidazol (benznidazol; Rochagan), que son los dos compuestos heterocíclicos comercializados en 1972 y 1974, respectivamente (1). Ninguno de los dos está aprobado por la FDA (Food and Drug Administration)(24) por lo que en Estados Unidos solo están disponibles a través de la CDC (Centers for Disease Control and Prevention) (25).
Desde entonces, se han preparado cientos de análogos correspondientes a diferentes familias, algunas de los cuales han mostrado interesantes propiedades anti-T. cruzi (Figura 3). Se pueden citar derivados de 5-nitrofurilo, 5-nitrotienilo; furoxanilo, benzofuroxanilo (26, 27); quinoxalinilo, imidazolilo o benzimidazolilo, indazolilo, (28), imidazolidinas (29), imidazo[4,5-c][1,2,6]tiadiazinas (30), imidazo[1,2-b]isoquinolinas (31). Otras familias descritas con propiedades anti-T. cruzi corresponden a N-óxidos de furoxano o benzofuroxano, imidazol o benzimidazol, indazol o quinoxalina (32, 33).
2.2. Diseño Basado en Cribado Sistemático
Este método consiste en someter a las nuevas moléculas objeto de estudio, bien sean de procedencia sintética o de origen natural, a evaluación farmacológica utilizando cualquier prueba biológica, sin una hipótesis previa. Se basa en el uso sistemático de baterías de test en cualquier modelo experimental, si bien la tendencia actual es utilizar pruebas in vitro, ensayos de anclaje (binding), inhibición enzimática, órganos aislados, cultivos celulares, etc. El cribado puede hacerse ensayando un pequeño número de moléculas, químicamente sofisticadas y novedosas, a una serie de pruebas biológicas o llevarse a cabo al azar buscando un hit entre centenares de moléculas. Una estrategia frecuentemente utilizada es la de incluir intermedios sintéticos, ya que en general, en las rutas de síntesis utilizadas para la síntesis de los compuestos objetivo, es necesario la preparación previa de productos intermedios que guardan una cierta similitud estructural con los compuestos finales.
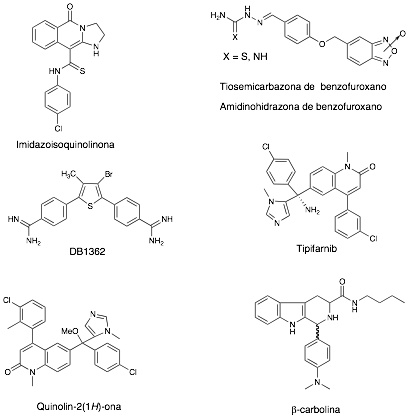
Figura 3.- Ejemplos representativos de derivados anti-T. cruzi.
Desde los años 80, con los avances de la robótica y de la miniaturización de los métodos de ensayos in vitro, en el HTS se han producido grandes avances con la introducción de estas mejoras. Este método se aplica utilizando técnicas de evaluación por desplazamiento de radioligandos o mediante procesos de inhibición enzimática. En la actualidad, la tendencia es sustituir los ensayos de radioligandos por ensayos de fluorescencia.
Las estrategias de screening que corresponden a métodos empíricos han sido las más utilizadas para el descubrimiento de nuevos prototipos en la búsqueda de compuestos anti- T. cruzi. Estas estrategias, aunque son aparentemente menos racionales, siguen siendo muy útiles para el descubrimiento de nuevos compuestos tripanomicidas, debido fundamentalmente al desarrollo y mejora de los ensayos in vitro en cepas de Tripanosoma cruzi.
El método de HTS que es ampliamente utilizado en la industria biofarmacéutica, ha sido poco aplicado en la búsqueda de nuevos fármacos para la enfermedad de Chagas. Uno de los más interesantes corresponde a un trabajo en el que utilizando una biblioteca química de 2000 compuestos, se describen 3 nuevos derivados (Figura 4) que inhiben la replicación intracelular de amastigotes en el rango nM, con una baja toxicidad (34).
2.3. Diseño Basado en Información Biológica
Esta estrategia se basa en la observación de efectos biológicos en humanos, en animales, en vegetales o en microorganismos. La procedencia de estos datos puede proceder de fuentes muy diversas como: 1) estudio de medicinas indígenas; 2) observaciones clínicas de los efectos adversos o secundarios de medicinas; 3) nuevos usos para medicamentos ya utilizados para otras dolencias; 4) observaciones fortuitas de moléculas usadas en la industria.
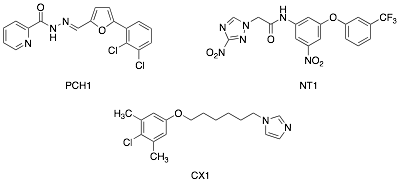
Figura 4.- Nuevos prototipos anti-T. cruzi descritos mediante un estudio HTS.
En este sentido, distintos principios activos han sido evaluados en este campo. Se pueden citar algunos que han sido estudiados sin buenos resultados, como los antibióticos, actinomicina D (35) o amfotericina B (36). Sin embargo, estudios en antifúngicos como el ketoconazol han demostrado efectividad en tejidos infectados en fase aguda, aunque con poca efectividad en fase crónica (37, 38). Mención aparte merece un análogo como el itraconazol (Figura 5), antifúngico perteneciente a la familia de los triazoles que está indicado en micosis superficiales y profundas. Este fármaco ejerce su efecto alterando la membrana celular del hongo mediante la inhibición de la síntesis del ergosterol interaccionando con la 14-a-desmetilasa, una enzima del citocromo P450 que es necesaria para la conversión del lanosterol a ergosterol. En el caso del itraconazol los estudios realizados en adultos demostraron una curación parasitológica en el 20% de los enfermos estudiados, con un 50% de mejoría de las alteraciones electrocardiográficas (39). Otro caso interesante es el alopurinol (inhibidor de la biosíntesis de purinas) que si bien no es eficaz en el tratamiento de pacientes en fase aguda si presenta efecto en pacientes crónicos (Figura 5). Estudios comparativos con benznidazol y nifurtimox, mostraron menos efectos adversos en el grupo tratado con alopurinol (39). Estos compuestos si bien han mostrado utilidad en algunos de los casos estudiados (25, 39-41) deben de ser objeto de más investigaciones con protocolos más rigurosos en un mayor número de pacientes.
2.4. Quimioinformática
La quimioinformática combina los recursos de la información, para transformar datos en información e información en conocimiento, con el objetivo de encontrar nuevos hits o para la optimización de los mismos, de manera más científica y racional, con el consiguiente ahorro de tiempo y de recursos económicos (42). Las estrategias generales para el diseño racional de fármacos se pueden plantear dependiendo del conocimiento o desconocimiento de la estructura tridimensional del receptor o diana.
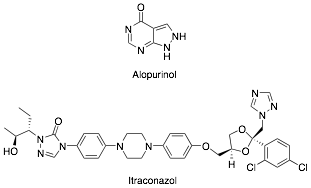
Figura 5.- Estructuras de medicamentos con actividad antichagásica.
2.4.1. Diseño basado en las moléculas bioactivas
Esta estrategia se basa en el estudio de moléculas cuyas propiedades farmacológicas han sido previamente demostradas y consiste en la búsqueda de elementos comunes, bien sean estructurales o relacionados con sus propiedades fisicoquímicas. Se pueden considerar diferentes modelos que se describen a continuación, dependiendo fundamentalmente de considerar propiedades 2D o 3D.
2.4.1.1. Modelo QSAR (relaciones estructura-actividad cuantitativa)
Para el establecimiento de un modelo QSAR, es necesario en primer lugar definir las moléculas mediante valores numéricos. Existen diferentes aproximaciones que se pueden considerar para la descripción de las estructuras bien sea mediante parámetros clásicos, definidos en función de criterios electrónicos, estéricos o hidrofóbicos o mediante descriptores que son calculados a partir de la representación de la estructura en dos o en tres dimensiones.
En general, los químicos representan las moléculas en dos dimensiones mediante un gráfico. Sin embargo, también pueden ser representadas por una serie de caracteres unidos y ordenados. Los más utilizados son el código SMILES (Simplified Molecular Input Line Entry System) o el InChI (International Chemical Identifier).
La representación de la estructura tridimensional de una molécula es mucho más compleja, si bien tanto la geometría como las propiedades asociadas puede ser establecida mediante cálculos de mecánica molecular o de mecánica cuántica (43-45) (métodos DFT, ab initio, semiempíricos). La elección de uno u otro método depende fundamentalmente del tamaño de la molécula, de la naturaleza del problema planteado y del tiempo de cálculo requerido.
Otro aspecto importante es el método matemático usado en la búsqueda de modelos de predicción. El método MLR (Multiple Lineal Regresion) (46, 47) que fue uno de los primeros en utilizarse a principios de los años noventa, permite encontrar relaciones lineales entre las propiedades observadas y un conjunto de descriptores. El principal problema radica en el hecho de que el número de moléculas usadas debe ser muy superior al número de descriptores empleados, y en que los descriptores o variables no independientes se traducen en modelos con una regresión de mala calidad.La regresión parcial por mínimos cuadrados o PLS (Partial Least Square) (47) soluciona en parte estos problemas, ya que es posible el uso de un número ilimitado de descriptores y soluciona el problema de la colinealidad entre variables.
Los modelos descritos en la bibliografía han sido utilizados para explicar la actividad en función de la estructura de series análogas o para el estudio de relaciones estructura actividad cuantitativa. En general, tienen más interés académico que utilidad para la búsqueda de nuevos prototipos estructuralmente diferentes. Los modelos publicados de estudios QSAR se han llevado a cabo con derivados de familias de semicarbazonas (48), imidazolinas (29), derivados heterocíclicos de N-óxido (32), 5-nitrofuril semicarbazonas (49), cumarinas (50), nitrofurazonas (51), etc utilizando la actividad anti-T. cruzi. En otros casos, la actividad antichagásica es representativa de un proceso enzimático como la inhibición de la Tripanotiona reductasa (51, 52) o de la cruzipaína (48, 53).
Estos modelos han utilizado diferentes tipos de descriptores que incluyen tanto parámetros clásicos, como otros obtenidos mediante química cuántica (29, 31, 54, 55). Las estrategias matemáticas más utilizados en el desarrollo de estos modelos corresponden a métodos de regresión lineal múltiple, si bien también han sido usados métodos no lineales (32, 48, 56). Un ejemplo que puede aplicarse para la predicción de nuevas moléculas (52), se refiere al desarrollo de un modelo cualitativo que mediante un análisis discriminante, permite diferenciar los compuestos activos/inactivos frente a la inhibición de la Tripanotiona reductasa (IC50) con un 90% y 89% de aciertos, en el conjunto de entrenamiento y validación, respectivamente. En el desarrollo del modelo se consideraron 1612 descriptores (geométricos y topológicos) y se estableció un modelo validado con 35 estructuras.
2.4.1.2. Redes neuronales artificiales
En el siglo XXI se ha incrementado la utilización de la inteligencia artificial, metodología basada en aproximaciones matemáticas no lineales. Este tipo de métodos consiguen simular procesos biológicos, ya sean cambios evolutivos o mutaciones en el caso de los algoritmos genéticos (57, 58) o el comportamiento de las redes neuronales biológicas en el caso de las RNA (redes neuronales artificiales) (59-62).
Una red neuronal artificial es un modelo de procesamiento de la información inspirado en el funcionamiento del sistema nervioso biológico y trata por tanto, de reproducir las características del cerebro humano. La estructura de este sistema de procesamiento se compone de un gran número de elementos interconectados (neuronas) que procesan la información recibida. El funcionamiento de una red neuronal depende de la arquitectura, del tipo de conexión entre las neuronas, y del mecanismo de aprendizaje (63) empleado en el proceso de entrenamiento.
Durante el proceso de aprendizaje la red neuronal puede modificar los pesos de las interconexiones en respuesta a los datos de entrada atendiendo a diferentes criterios (64). En el aprendizaje supervisado el proceso se realiza mediante un entrenamiento controlado por un agente externo (supervisor o maestro) que determina la respuesta que debería generar la red a partir de los datos de entrada. En el aprendizaje no supervisado, la red no requiere de la influencia externa para ajustar los pesos de las conexiones entre sus neuronas y por tanto, no recibe ninguna información por parte del entorno que le indique si la salida generada en respuesta a una entrada es o no correcta. Este tipo de redes son capaces de autoorganizarse, buscando las características, regularidades o patrones que se pueden establecer entre los datos de entrada.
Un ejemplo de aplicación de esta metodología ha sido realizada en 46 cetonas en las que se ha establecido un modelo de correlación con la constante de inhibición de la cruzipaína mediante una red neuronal siguiendo el método de regulación Bayesiana (65).
Otra estrategia diferente basada en redes neuronales artificiales que permite la predicción de la actividad anti-T. cruzi de cualquier tipo de molécula ha sido descrita por nuestro grupo de trabajo (66). Este procedimiento está basado íntegramente en la utilización de redes neuronales artificiales tanto en el proceso de definición de las moléculas como en el modelo de predicción. La metodología desarrollada para la obtención del modelo matemático, consiste en varias etapas (Figura 6).
Un conjunto representativo de 72 compuestos obtenidos de Cerecetto y col. (33) con diversidad tanto estructural como biológica fue seleccionado para el estudio. Teniendo en cuenta el porcentaje de la inhibición del crecimiento, la definición de las moléculas fue realizada mediante una red neuronal no supervisada utilizando el programa CODES® (67). CODESÒ genera descriptores topológicos de las estructuras mediante una red neuronal basada en un modelo de activación y competición interactiva (IAC) a partir de las estructuras graficas. CODESÒ parte de la representación gráfica de la molécula, generando el correspondiente espacio topológico que posteriormente es trasladado a un espacio neuronal (red neuronal isomorfa). Para cada molécula, CODESÒ genera un vector numérico con tantos elementos (descriptores) como átomos tenga la molécula. Puesto que el procesamiento de esta RNA es secuencial, para cada subproceso o iteración se generará un vector de actividad. El conjunto de todos estos vectores constituye una matriz dinámica, con tantas columnas como átomos tenga la molécula y cuyo último vector representa el estado de equilibrio del proceso.
La reducción de dimensiones de cada matriz dinámica (las estructuras quedan codificadas en cuatro descriptores) se realiza mediante una red neuronal de retropropagación del error y es necesaria para no perder la información de la matriz dinámica de CODES y para que el número de variables sea el mismo para todas las moléculas. Durante el entrenamiento de la red se estudian diferentes arquitecturas utilizando una red de retropropagación del error mediante un aprendizaje supervisado, ya que cada molécula de la serie de entrenamiento va asociada con su valor de actividad.
Siguiendo esta metodología se ha desarrollado una red neuronal (68) de arquitectura 4-4-1, a partir de 37 compuestos que presenta una capacidad de predicción del 78% en el test de validación externa (35 compuestos) (Figura 7).
Las ventajas más destacables de esta metodología son: a) se puede aplicar a conjuntos de moléculas quimio- y biodiversas; b) no es necesario conocer la estructura tridimensional de los mínimos conformacionales; c) no es necesaria una selección previa de parámetros para definir las moléculas; d) es aplicable a moléculas quirales.
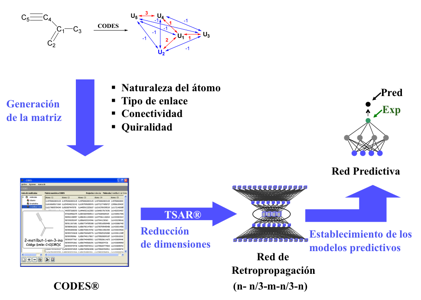
Figura 6.- Procedimiento para el establecimiento de una red neuronal para la predicción de la actividad antichagásica.
Esta metodología proporciona una herramienta de gran utilidad ya que la red final es capaz de estimar la inhibición del crecimiento parasitario de familias estructurales quimiodiversas de manera rápida y eficaz, lo que permite evitar costes innecesarios en el desarrollo de nuevos compuestos con potencialidad terapéutica.
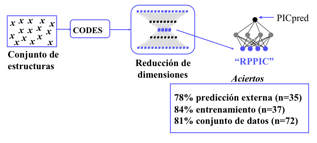
Figura 7.- Esquema de la red neuronal para la predicción de la actividad anti-T. cruzi (porcentaje de inhibición del crecimiento del parásito, PIC).
2.4.1.3. Modelo de farmacóforo
Los requerimientos estéricos y electrónicos mínimos indispensables de una molécula para lograr la unión al receptor y originar una respuesta farmacológica es lo que constituye el concepto de farmacóforo (69) (Figura 8). Sin embargo, la presencia de un farmacóforo en cualquier estructura química no es condición suficiente para esperar una respuesta farmacológica.
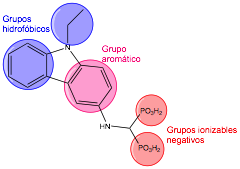
Figura 8.- Farmacóforo de los inhibidores de TcHx (adaptado de (70)).
El procedimiento seguido para el diseño de un farmacóforo comienza con la búsqueda de la conformación bioactiva, mediante técnicas de exploración del espacio conformacional. El segundo paso consiste en un proceso de superposición estructural de las moléculas consideradas. Y por ultimo, el establecimiento de los requerimientos mínimos que tiene que cumplir una molécula para interaccionar con la diana biológica propuesta. Este tipo de estudio necesita de un análisis conformacional riguroso de cada una de las moléculas a analizar, porque es posible que la conformación bioactiva del fármaco no sea la más estable termodinámicamente. Algunos de los programas más utilizados en la elucidación del farmacóforo son Catalyst (71), Galahad (72) y Gasp (73).
2.4.1.4. Modelo 3D-QSAR
El 3D-QSAR considera la naturaleza tridimensional de las moléculas, asumiendo que la interacción molécula-receptor puede ser representada por un conjunto de interacciones estéricas y electrostáticas, entre la molécula a estudiar y un “átomo sonda”. Estas interacciones se conocen como MIP (Molecular Interaction Potential). Estadísticamente, no se trata de un modelo lineal como en el dimensional ya que vamos a tener una gran cantidad de variables, por tanto no se puede utilizar como herramienta estadística una regresión, sino que es necesario aplicar otras técnicas que permitan disminuir el número de variables (PLS).
Los métodos 3D-QSAR más utilizados son CoMFA (74, 75), CoMSIA (76, 77) y GRID/GOLPE (78). La principal diferencia entre estos métodos es el tipo de descriptores que usan: los métodos CoMFA y CoMSIA calculan diferentes campos de interacción molecular, estéricos, electrostáticos, hidrofóbicos. El método GRID/GOLPE permite calcular un campo para cada sonda química implementada en el programa GRID, que utiliza aproximaciones de mecánica molecular.
Un ejemplo interesante de la aplicación de estas técnicas se puede encontrar en el diseño de inhibidores de la enzima hexoquinasa (HK) (70). La hexoquinasa es la primera enzima involucrada en la glicólisis de muchos organismos, entre los que se incluye el agente etiológico de la enfermedad de Chagas, T. cruzi.
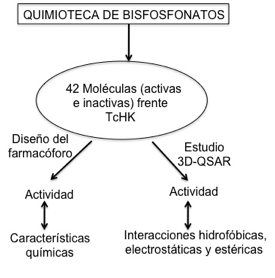
Figura 9.- Procedimiento empleado para el desarrollo de bisfosfonatos como inhibidores de la enzima hexoquinasa de T. cruzi (TcHK).
En base a estudios previos que mostraban que análogos de los bisfosfonatos eran potentes inhibidores de la hexoquinasa de T. cruzi, y haciendo uso de las herramientas antes comentadas como la construcción de un modelo de farmacóforo y estudios 3D-QSAR (CoMSIA) se ha explicado la actividad de los derivados de bisfosfonatos frente a la enzima hexoquinasa de T. cruzi y se han diseñado nuevos análogos (Figuras 9 y 10).
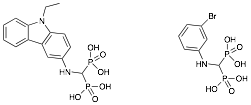
Figura 10.- Ejemplo de bisfosfonatos estudiados.
El modelo de farmacóforo construido con 17 compuestos activos considerando hasta un máximo de 256 conformaciones por cada compuesto, permitió confirmar la hipótesis de partida donde se presuponía que se necesitaban 2 grupos negativos ionizables, 2 grupos hidrofóbicos y un grupo aromático neutro (Figura 11).
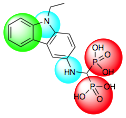
Figura 11.- Modelo de farmacóforo para la inhibición de TcHK.
El estudio 3D-QSAR se llevó a cabo con el programa CoMSIA, que calcula índices de similitud basándose en la interacciones (hidrofóbicas, electrostáticas y estéricas) entre una molécula y un átomo prueba, para posteriormente correlacionar dichos índices con la actividad. Los pasos seguidos en el estudio tridimensional se pueden resumir en los siguientes puntos: i) alineamiento del grupo de entrenamiento usando el patrón (H)O-PC-P-O(H); ii) cálculo de las interacciones de cada molécula del conjunto de entrenamiento con un átomo prueba (átomo sp3 C); iii) análisis cuantitativo empleando PLS. Los resultados derivados del estudio 3D-QSAR (CoMSIA) pusieron de manifiesto que la contribución electrostática es más importante que la hidrofóbica o la estérica (43% frente al 24% y 13 % respectivamente) (Figura 12).
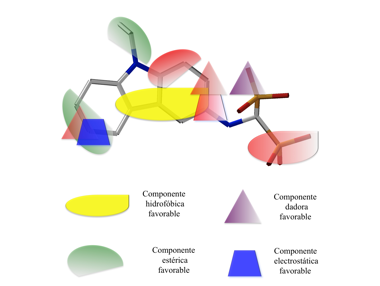
Figura 12.- Esquema de la contribución de las diferentes interacciones. El color rojo indica interacciones desfavorables.
De acuerdo con los resultados obtenidos se puede explicar por qué algunos bisfosfonatos que inhiben la enzima farnesil difosfato sintetasa (FPPS), no son inhibidores de la enzima TcHK. Para que un compuesto cumpla las condiciones en el modelo de farmacóforo de los inhibidores de FPPS es necesario una carga positiva, mientras que en el modelo de farmacóforo de los inhibidores de TcHK los puntos claves son grupos voluminosos y cadenas no cargadas (Figura 12 ).
2.4.2. Diseño basado en la estructura tridimensional de la diana. Estudio de las interacciones fármaco-receptor
Una gran variedad de procesos fisiológicos son la consecuencia de la interacción entre ligandos y macromoléculas. Conociendo la estructura de una determinada proteína es posible encontrar ligandos que puedan modificar la función de dicha proteína. Estas dianas pueden ser enzimas, receptores, canales de iones, proteínas de transporte o DNA. En general los fármacos que interaccionan en estas dianas actúan mediante procesos de: inhibición enzimática, interferencia en la biosíntesis de la pared molecular, alteración del transporte en membranas celulares o mediante interacción con receptores de membrana o con receptores intracelulares.
La caracterización de la secuencia del genoma del T. cruzi que fue publicada en 2005 (79) y el correspondiente análisis proteómico, ha permitido la identificación de características especificas de las diferentes etapas de su ciclo de vida (80). Desde entonces, el progreso en los conocimientos sobre su fisiología, bioquímica, nuevas rutas biosintéticas y las enzimas relevantes para la supervivencia del T. cruzi han sido muy relevantes y han proporcionado nuevas dianas para la exploración de nuevas estrategias para la quimioterapia en la enfermedad de Chagas.
Varias estructura terciarias obtenidas mediante cristalografía de rayos X de enzimas de T. cruzi han sido descritas y almacenadas en la base de datos de proteínas, PDB (protein data bank) (81). En la Tabla 1 se recogen las enzimas más representativas que podrían ser utilizadas en el diseño de nuevos fármacos contra la enfermedad de Chagas.
Tabla 1.- Dianas representativas con estructura tridimensional.
Diana |
Estructuras cristalográficas |
Mecanismo de acción |
Hipoxantina fosforibosil- transferasa (HGPRT) |
1P17, 1P18, 1P19, 1I0I, 1I0L, 1I13, 1IL14, 1TC2, 1TC1 |
Captura de purinas y |
Dihidrofolato reductasa– Timidilato sintetasa (DHFR-TS) |
síntesis de nucleótidos |
|
Cruzipaína |
1AIM, 1EWL, 1EWM, 1EWO, 1EWP, 1F29, 1F2A, 1F2B, 1F2C, 1ME3, 1ME4, 1U9Q, 2AIM, 2EFM, 2OUL, 2OZ2 |
Responsable de la actividad proteolítica |
Tripanotiona reductasa (TR) |
1GXF, 1AOG, 1NDA |
Síntesis y metabolismo del tripanotiona |
Farnsesil pirofosfato Sintetasa (FPPS) |
3IBA, EICK, 3ICM, 3ICN, 3ICZ, 3ID0, 1YHK, 1YHL, 1YHM |
Biosíntesis de ergosterol |
Transialidasa |
1MR5, 1MS0, 1MS1, 1MS3, 1MS4, 1MS5, 1MS8, 1MS9, 1S0I, 1S0J, 2AH2 |
Transferencia de residuos de ácido siálico |
Gliceraldehido-3-fosfato deshidrogenasa (GAPDH) |
Vía metabólica de oxidación de la glucosa |
|
C14α esterol demetilasa |
2WUZ, 2WXZ |
Biosíntesis de esteroles de membrana |
2.4.2.2. Estudios de Anclaje
Los estudios de anclaje (docking) son una herramienta computacional muy útil para buscar la mejor unión entre dos moléculas, un receptor y un ligando, con el fin de diseñar ligandos más específicos que encajen con mayor precisión en el sitio de unión de la diana terapéutica (82-84).
En función de la información experimental de la que se disponga, la predicción de la unión proteína-ligando puede ser más o menos costosa. Si se dispone de complejos cristalizados ligando-receptor, el estudio del sitio de unión de nuevas moléculas resulta mucho más sencillo. Por otra parte, poder disponer de datos biológicos experimentales como los obtenidos por mutagénesis dirigida, permiten deducir las interacciones entre el fármaco y la proteína. Con estas informaciones, se puede realizar un acoplamiento manual entre el ligando y el receptor mediante programas de modelización molecular para obtener así modelos que expliquen en lo posible los datos experimentales. Sin embargo, si no se dispone de datos experimentales (complejo 3D proteína-ligando cristalizado, estudios de mutagénesis, etc), existen métodos automáticos para explorar las posibles uniones entre ligando y receptor. Son los denominados programas de docking, los cuales realizan una exploración de todas las posibles posiciones relativas ligando-receptor evaluando la interacción intermolecular entre ambos mediante una función de scoring (85,86)
Los métodos más utilizados son: i) fast shape matching (DOCK(87), EUDOCK(88), LIGANDFIT(89)), ii) construcción incremental del ligando en la cavidad de la proteína (FLEXX(90)) y iii) algoritmos genéticos (FLEXIDOCK, GOLD(91), AUTODOCK3.0(92)),
Las aplicaciones de cribado virtual (93) permiten, a partir de grandes bases de datos de estructuras químicas (“quimiotecas” o chemical libraries), seleccionar aquellos compuestos que presentan una mayor afinidad por la diana terapéutica. En el cribado virtual se emplean diferentes filtros para ir reduciendo el número de estructuras. Estos filtros pueden realizarse empleando descriptores moleculares o definiendo un modelo de farmacóforo.. Se requiere poseer un archivo numeroso de ligandos potenciales, esto es, moléculas orgánicas con estructuras tridimensionales conocidas.
Una de las quimiotecas más utilizada es ZINC, que es una base de datos de libre distribución (94) cuyo catálogo completo (2.8*106 moléculas) incluye moléculas de varias casas comerciales.
Un ejemplo interesante de la utilización de estos métodos se aplicó en el diseñó de inhibidores de la enzima hipoxantina-guanina fosforibosil transferasa (HGPRT)(98). El Tripanosoma cruzi es deficiente en la síntesis de novo de las purinas, por lo que deben obtener estos compuestos esenciales del hospedero. Una enzima esencial en este proceso es la hipoxantina-guanina fosforibosil transferasa (HGPRT). Diferentes estructuras tridimensionales de esta enzima (95-97) han sido resueltas mediante cristalografía de rayos X (Tabla 1). Gracias al conocimiento de estas estructuras junto con estudios de mutaciones se ha podido describir el sitio activo en el bucle I, definido por tres residuos análogos en la enzima humana, Leu67, Lys68 y Gly69. Mediante cribado virtual y usando la estructura cristalográfica de la HGPRT (Código PDB 1TC2) se identificaron 22 potenciales inhibidores, 16 de los cuales demostraron ser potentes inhibidores de la HGPRT y 3 de ellos efectivos agentes antiproliferativos in vitro contra la forma amastigote intracelular del parásito (Figura 13). El cribado virtual se realizó empleando un programa de docking flexible (DOCK), enfrentando el sitio activo de la enzima HGPRT frente a la base de datos ACD (Available Chemicals Directory de MDL Information Systems, San Leandro, CA, USA).
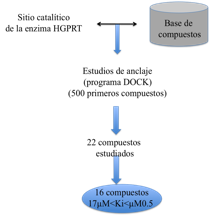
Figura 13.- Estrategia empleada en la identificación de nuevos inhibidores de HGPRT.
Otro ejemplo muy interesante de la aplicación de las técnicas de docking y cribado virtual al desarrollo de nuevos inhibidores de la enzima cruzipaína ha sido publicado recientemente (99).
La cruzipaína, una cisteín-proteasa, es responsable de la mayor parte de la actividad proteolítica de T. cruzi en todos los estadios de su ciclo de vida. Inhibidores selectivos de esta proteasa son capaces de bloquear la proliferación tanto de la forma extracelular (epimastigotes) como de los amastigotes intracelulares, así como de impedir la transformación de epimastigotes a tripomastigotes metacíclicos, lo que indica que la proteína tiene funciones esenciales en el ciclo de vida del parásito. Ferreira y colaboradores (99) emplean de forma complementaria el cribado virtual (CV) y el cribado de alto rendimiento (HTS, high-throughput screening) de una quimioteca en busca de inhibidores reversibles y competitivos de la enzima cruzipaína (Figura 14).
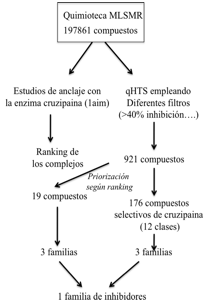
Figura 14.- Estrategia combinada de HTS y CV para el desarrollo de inhibidores competitivos de la enzima cruzipaína.
3. CONCLUSIONES
La singularidad de la enfermedad de Chagas, como enfermedad íntimamente relacionada con la pobreza y la ausencia de salubridad han sido, entre otros, factores fundamentales en el deficiente desarrollo de nuevos medicamentos. En la actualidad se siguen utilizando fármacos que fueron comercializados en los años 70, como el nifurtimox y el benznidazol que no están aprobados por la FDA. El hecho es que esta dolencia está considerada por la Organización Mundial de la Salud como una enfermedad extremadamente olvidada.
La situación terapéutica en la enfermedad de Chagas no puede explicarse de ninguna manera, ni en función del estado actual del conocimiento de la enfermedad, ni de las herramientas disponibles para el diseño de fármacos, ya que son comparables con las existentes para cualquier otra enfermedad. Parece por tanto, que aunque el diseño racional es siempre útil y recomendable para que el descubrimiento de nuevos fármacos sea sostenible, se hace más indispensable, aún si cabe, para la búsqueda de nuevos medicamentos eficaces para la enfermedad de Chagas.
Es por tanto conveniente y deseable la utilización de todos los métodos disponibles independientemente que sean más o menos clásicos o que estén basados en técnicas más novedosas como las relacionadas con la quimioinformática. En este sentido, la caracterización de la secuencia del genoma del Tripanosoma cruzi en 2005 y el correspondiente análisis proteómico, ha puesto al descubierto nuevas rutas biosintéticas y proteínas relevantes para la supervivencia del parásito y ha abierto nuevas líneas de investigación. En este sentido, en el desarrollo de nuevos fármacos aplicando métodos quimioinformáticos hay que resaltar entre otros, los que se refieren a los inhibidores de la hipoxantina-guanina fosforribosil transferasa, de la cruzipaína y de la sintetasa de la farnesil-pirofosfato o hexoquinasa.
Con todos los recursos científicos disponibles, el descubrimiento de nuevos cabezas de serie (lead compound) y de nuevos fármacos debería de ser un proceso más rápido, económico, eficiente y sostenible.
4. AGRADECIMIENTOS
Los autores quieren agradecer al Ministerio de Ciencia e Innovación (proyecto TRA2009_0085, 2010-2011), al CSIC (proyecto de cooperación 2009UY01) y a la red CYTED (http://ridimedchag.fq.edu.uy; RIDIMEDCHAG) la ayuda recibida.
5. REFERENCIAS
1. Sanchez-Sancho, F., Campillo, N. E., & Paez, J. A. (2010) Chagas disease: progress and new perspectives. Curr Med Chem 17(5), 423-452
2. Pan American Health Organization (PAHO: http://www.paho.org).
3. Global Plan To Combat Neglected Tropical Diseases 2008–2015. World Health Organization 2007.
4. Wermuth, C. G. (2008). The Practice of Medicinal Chemistry. 3 edn. Elsevier Ltd.: Madrid.
5. Avendaño, C. (2001). Introducción a la Química Farmacéutica. Vol. 1, 2ª edn. Mc Graw Hill: Madrid.
6. Sepulveda-Boza, S., & Cassels, B. K. (1996). Plant metabolites active against Trypanosoma cruzi. Planta Med 62(2), 98-105.
7. Coura, J. R., & de Castro, S. L. (2002). A critical review on Chagas disease chemotherapy. Mem Inst Oswaldo Cruz 97(1), 3-24.
8. Maya, J. D., Cassels, B. K., Iturriaga-Vasquez, P., Ferreira, J., Faundez, M., Galanti, N., Ferreira, A., & Morello, A. (2007). Mode of action of natural and synthetic drugs against Trypanosoma cruzi and their interaction with the mammalian host. Comp Biochem Physiol A Mol Integr Physiol 146(4), 601-620.
9. Izumi, E., Ueda-Nakamura, T., Dias Filho, B. P., Veiga Junior, V. F., & Nakamura, C. V. (2011). Natural products and Chagas' disease: a review of plant compounds studied for activity against Trypanosoma cruzi. Nat Prod Rep 28(4), 809-823.
10. Menna-Barreto, R. F., Laranja, G. A., Silva, M. C., Coelho, M. G., Paes, M. C., Oliveira, M. M., & de Castro, S. L. (2008). Anti-Trypanosoma cruzi activity of Pterodon pubescens seed oil: geranylgeraniol as the major bioactive component. Parasitol Res 103(1), 111-117.
11. Reyes-Chilpa, R., Estrada-Muniz, E., Vega-Avila, E., Abe, F., Kinjo, J., & Hernandez-Ortega, S. (2008). Trypanocidal constituents in plants: 7. Mammea-type coumarins. Mem Inst Oswaldo Cruz 103(5), 431-436.
12. Flores, N., Jimenez, I. A., Gimenez, A., Ruiz, G., Gutierrez, D., Bourdy, G., & Bazzocchi, I. L. (2008). Benzoic acid derivatives from Piper species and their antiparasitic activity. J Nat Prod 71(9), 1538-1543.
13. Batista, J. M., Jr., Lopes, A. A., Ambrosio, D. L., Regasini, L. O., Kato, M. J., Bolzani Vda, S., Cicarelli, R. M., & Furlan, M. (2008). Natural chromenes and chromene derivatives as potential anti-trypanosomal agents. Biol Pharm Bull 31(3), 538-540.
14. Lopes, A. A., Lopez, S. N., Regasini, L. O., Junior, J. M., Ambrosio, D. L., Kato, M. J., da Silva Bolzani, V., Cicarelli, R. M., & Furlan, M. (2008). In vitro activity of compounds isolated from Piper crassinervium against Trypanosoma cruzi. Nat Prod Res 22(12), 1040-1046.
15. Felippe, L. G., Baldoqui, D. C., Kato, M. J., Bolzani Vda, S., Guimaraes, E. F., Cicarelli, R. M., & Furlan, M. (2008). Trypanocidal tetrahydrofuran lignans from Peperomia blanda. Phytochemistry 69(2), 445-450.
16. Vik, A., Proszenyak, A., Vermeersch, M., Cos, P., Maes, L., & Gundersen, L. L. (2009). Screening of agelasine D and analogs for inhibitory activity against pathogenic protozoa; identification of hits for visceral leishmaniasis and Chagas disease. Molecules 14(1), 279-288.
17. Rosa, L. H., Machado, K. M., Rabello, A. L., Souza-Fagundes, E. M., Correa-Oliveira, R., Rosa, C. A., & Zani, C. L. (2009). Cytotoxic, immunosuppressive, trypanocidal and antileishmanial activities of Basidiomycota fungi present in Atlantic Rainforest in Brazil. Antonie Van Leeuwenhoek 95(3), 227-237.
18. Abe, F., Nagafuji, S., Yamauchi, T., Okabe, H., Maki, J., Higo, H., Akahane, H., Aguilar, A., Jimenez-Estrada, M., & Reyes-Chilpa, R. (2002). Trypanocidal constituents in plants 1. Evaluation of some Mexican plants for their trypanocidal activity and active constituents in Guaco, roots of Aristolochia taliscana. Biol Pharm Bull 25(9), 1188-1191.
19. Abegaz, B. M., Bezabih, M., Msuta, T., Brun, R., Menche, D., Muhlbacher, J., & Bringmann, G. (2002). Gaboroquinones A and B and 4'-O-demethylknipholone-4'-O-beta-D-glucopyranoside, phenylanthraquinones from the roots of Bulbine frutescens. J Nat Prod 65(8), 1117-1121.
20. Ambrosio, S. R., Arakawa, N. S., Esperandim, V. R., de Albuquerque, S., & Da Costa, F. B. (2008). Trypanocidal activity of pimarane diterpenes from Viguiera arenaria (Asteraceae). Phytother Res 22(10), 1413-1415.
21. Sartorelli, P., Carvalho, C. S., Reimao, J. Q., Ferreira, M. J., & Tempone, A. G. (2009). Antiparasitic activity of biochanin A, an isolated isoflavone from fruits of Cassia fistula (Leguminosae). Parasitol Res 104(2), 311-314.
22. Vieira, N. C., Espindola, L. S., Santana, J. M., Veras, M. L., Pessoa, O. D., Pinheiro, S. M., de Araujo, R. M., Lima, M. A., & Silveira, E. R. (2008). Trypanocidal activity of a new pterocarpan and other secondary metabolites of plants from Northeastern Brazil flora. Bioorg Med Chem 16(4), 1676-1682.
23. Chagas, C. (1909). Nova tripanozomiase humana. Estudos sobre a morfologia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., agente etiológico de nova entidade mórbida do homem. Mem Inst Oswaldo Cruz 1: 159-218.
24. Food and Drug Administration (FDA). http://www.fda.gov.
25. Center for Disease Control and Prevention. Division of Parasitic Diseases (DPD), Antiparasitic treatment of Chagas disease Available at. http://www.cdc.gov/chagas/hcp/tx.html.
26. Porcal, W., Hernandez, P., Boiani, L., Boiani, M., Ferreira, A., Chidichimo, A., Cazzulo, J. J., Olea-Azar, C., Gonzalez, M., & Cerecetto, H. (2008). New trypanocidal hybrid compounds from the association of hydrazone moieties and benzofuroxan heterocycle. Bioorg Med Chem 16(14), 6995-7004.
27. Rodriguez, J., Gerpe, A., Aguirre, G., Kemmerling, U., Piro, O. E., Aran, V. J., Maya, J. D., Olea-Azar, C., Gonzalez, M., & Cerecetto, H. (2009). Study of 5-nitroindazoles' anti-Trypanosoma cruzi mode of action: electrochemical behaviour and ESR spectroscopic studies. Eur J Med Chem 44(4), 1545-1553.
28. Boiani, L., Gerpe, A., Aran, V. J., Torres de Ortiz, S., Serna, E., Vera de Bilbao, N., Sanabria, L., Yaluff, G., Nakayama, H., Rojas de Arias, A., Maya, J. D., Morello, J. A., Cerecetto, H., & Gonzalez, M. (2009). In vitro and in vivo antitrypanosomatid activity of 5-nitroindazoles. Eur J Med Chem 44(3), 1034-1040.
29. Caterina, M. C., Perillo, I. A., Boiani, L., Pezaroglo, H., Cerecetto, H., Gonzalez, M., & Salerno, A. (2008). Imidazolidines as new anti-Trypanosoma cruzi agents: biological evaluation and structure-activity relationships. Bioorg Med Chem 16(5), 2226-2234.
30. Paéz, J. A., Campillo, N. E., Guerra, A., Gonzalez, M., & Cerecetto, H. Una Nueva Familia de Antichagásicos Derivados de 2,2-Dióxido de Imidazo[4,5-c][1,2,6]tiadiazina. Patente ESP 200900225; PCT1641-296. 2010 Ene 26.
31. Bollini, M., Casal, J. J., Alvarez, D. E., Boiani, L., Gonzalez, M., Cerecetto, H., & Bruno, A. M. (2009). New potent imidazoisoquinolinone derivatives as anti-Trypanosoma cruzi agents: biological evaluation and structure-activity relationships. Bioorg Med Chem 17(4), 1437-1444.
32. Boiani, M., Cerecetto, H., Gonzalez, M., & Gasteiger, J. (2008). Modeling anti-Trypanosoma cruzi activity of N-oxide containing heterocycles. J Chem Inf Model 48(1), 213-219.
33. Cerecetto, H., & Gonzalez, M. (2008). Anti-T. cruzi agents: our experience in the evaluation of more than five hundred compounds. Mini Rev Med Chem 8(13), 1355-1383.
34. Bettiol, E., Samanovic, M., Murkin, A. S., Raper, J., Buckner, F., & Rodriguez, A. (2009). Identification of Three Classes of Heteroaromatic Compounds with Activity against Intracellular Trypanosoma cruzi by Chemical Library Screening. PLoS Negl Trop Dis 3(2), e384.
35. da Cruz, M. Q., Brascher, H. M., Vargens, J. R., & Oliveira-Lima, A. (1991). Effect of actinomycin D on Trypanosoma cruzi. Experientia 47(1), 89-92.
36. Cruz, F. S., Marr, J. J., & Berens, R. L. (1980). Prevention of transfusion-induced Chagas' disease by amphotericin B. Am J Trop Med Hyg 29(5), 761-765.
37. McCabe, R. (1988). Failure of ketoconazole to cure chronic murine Chagas' disease. J Infect Dis 158(6), 1408-1409.
38. McCabe, R. E., Remington, J. S., & Araujo, F. G. (1984). Ketoconazole inhibition of intracellular multiplication of Trypanosoma cruzi and protection of mice against lethal infection with the organism. J Infect Dis 150(4), 594-601.
39. Apt, W., Heitmann, I., Jercic, I., Jofré, L., Muñoz, P., Noemí, I., San Martín, A. M., Sapunar, J., Torres, M., & Zulantay, I. (2008). [Part VI. Antiparasitic treatment for Chagas disease]. Rev Chilena Infectol 25(5), 384-389, discussion 387.
40. Apt, W., Arribada, A., Zulantay, I., Solari, A., Sanchez, G., Mundaca, K., Coronado, X., Rodriguez, J., Gil, L. C., & Osuna, A. (2005). Itraconazole or allopurinol in the treatment of chronic American trypanosomiasis: the results of clinical and parasitological examinations 11 years post-treatment. Ann Trop Med Parasitol 99(8), 733-741.
41. Zulantay, I., Apt, W., Rodriguez, J., Venegas, J., & Sanchez, G. (1998). [Serologic evaluation of treatment of chronic Chagas disease with allopurinol and itraconazole]. Rev Med Chil 126(3), 265-270.
42. Oprea, T., Mannhold, R., Kubinyi, H., & Folkers, G. (2006). Chemoinformatics in Drug Discovery. Wiley-VCH.
43. Koch, W., & Holthausen, M. C. (2001). A Chemist’s Guide to Density Functional Theory. 3ª edn. Wiley-VCH Verlag GmbH.
44. Young, D. C. (2001). Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems. 3ª edn. John Wiley & Sons, Inc.
45. Hehre, W. S., Radom, L., Schleyer, P. v. R., & Pople, J. A. (1986). Ab initio Molecular Orbital Theory. 3ª edn. John Wiley & Sons, Inc.
46. Freund, R., & Wilson, W. (1998). Regression Analysis: Statistical Modeling of a Response Variable. Academic Press.
47. Draper, N., & Smith, H. (1998). Applied regression analysis. Wiley New York.
48. Guido, R. V., Trossini, G. H., Castilho, M. S., Oliva, G., Ferreira, E. I., & Andricopulo, A. D. (2008). Structure-activity relationships for a class of selective inhibitors of the major cysteine protease from Trypanosoma cruzi. J Enzyme Inhib Med Chem 23(6), 964-973.
49. Aguirre, G., Boiani, M., Cabrera, E., Cerecetto, H., Di Maio, R., Gonzalez, M., Denicola, A., Sant'anna, C. M., & Barreiro, E. J. (2006). New potent 5-nitrofuryl derivatives as inhibitors of Trypanosoma cruzi growth. 3D-QSAR (CoMFA). studies. Eur J Med Chem 41(4), 457-466.
50. Menezes, I. R., Lopes, J. C., Montanari, C. A., Oliva, G., Pavao, F., Castilho, M. S., Vieira, P. C., & Pupo, M. T. (2003). 3D QSAR studies on binding affinities of coumarin natural products for glycosomal GAPDH of Trypanosoma cruzi. J Comput Aided Mol Des 17(5-6), 277-290.
51. Martinez-Merino, V., & Cerecetto, H. (2001). CoMFA-SIMCA model for antichagasic nitrofurazone derivatives. Bioorg Med Chem 9(4), 1025-1030.
52. Prieto, J. J., Talevi, A., & Bruno-Blanch, L. E. (2006). Application of linear discriminant analysis in the virtual screening of antichagasic drugs through trypanothione reductase inhibition. Mol Divers 10(3), 361-375.
53. Trossini, G. H., Guido, R. V., Oliva, G., Ferreira, E. I., & Andricopulo, A. D. (2009). Quantitative structure-activity relationships for a series of inhibitors of cruzain from Trypanosoma cruzi: Molecular modeling, CoMFA and CoMSIA studies. J Mol Graph Model.
54. Paulino, M., Alvareda, E. M., Denis, P. A., Barreiro, E. J., Sperandio da Silva, G. M., Dubin, M., Gastellu, C., Aguilera, S., & Tapia, O. (2008). Studies of trypanocidal (inhibitory). power of naphthoquinones: evaluation of quantum chemical molecular descriptors for structure-activity relationships. Eur J Med Chem 43(10), 2238-2246.
55. Vera-Divaio, M. A., Freitas, A. C., Castro, H. C., de Albuquerque, S., Cabral, L. M., Rodrigues, C. R., Albuquerque, M. G., Martins, R. C., Henriques, M. G., & Dias, L. R. (2009). Synthesis, antichagasic in vitro evaluation, cytotoxicity assays, molecular modeling and SAR/QSAR studies of a 2-phenyl-3-(1-phenyl-1H-pyrazol-4-yl)-acrylic acid benzylidene-carbohydrazide series. Bioorg Med Chem 17(1), 295-302.
56. Freitas, R. F., Oprea, T. I., & Montanari, C. A. (2008). 2D QSAR and similarity studies on cruzain inhibitors aimed at improving selectivity over cathepsin L. Bioorg Med Chem 16(2), 838-853.
57. Rogers, D., & Hopfinger, A. (1994). Application of Genetic Function Approximation to Quantitative Structure-Activity Relationships and Quantitative Structure-Property Relationships. J Chem Inf Model 34(4), 854-866.
58. Devillers, J. (1996a). Genetic Algorithms in Molecular Modeling. Academic Press.
59. Livingstone, D. J., Manallack, D. T., & Tetko, I. V. (1997). Data modelling with neural networks: advantages and limitations. J Comput Aided Mol Des 11(2), 135-142.
60. Livingstone, D. J., & Manallack, D. T. (1993). Statistics using neural networks: chance effects. J Med Chem 36(9), 1295-1297.
61. Devillers, J. (1996b). Neural Networks in QSAR and Drug Design (Principles of QSAR and Drug Design). 1st edn. Academic Press: Lyon.
62. Zupan, J., & Gasteiger, J. (1999). Neural networks in chemistry and drug desing. 2nd edn. WILEY-VCH: Weinheim.
63. Kaiser, M. (2007). Brain architecture: a design for natural computation. Philos Transact A Math Phys Eng Sci 365(1861), 3033-3045.
64. Ichikawa, H. (2003). Hierarchy neural networks as applied to pharmaceutical problems. Adv Drug Deliv Rev 55(9), 1119-1147.
65. Caballero, J., Tundidor-Camba, A., & Fernaández, M. (2007). Modeling of the Inhibition Constant (Ki). of Some Cruzain Ketone-Based Inhibitors Using 2D Spatial Autocorrelation Vectors and Data-Diverse Ensembles of Bayesian Regularized Genetic Neural Networks QSAR & Comb Sci 26(1), 27-40.
66. Paéz, J. A., Campillo, N. E., Guerra, A., Gonzalez, M., & Cerecetto, H. (2009). Procedimiento para la determinación de la actividad antichagásica de una molécula. Patente ESP P200900242.
67. M. Stud CODES©; v1.0 (revision 3). ed.
68. Paéz, J. A., Campillo, N. E., Guerra, A., Gonzalez, M., & Cerecetto, H. (2009). Procedimiento para la determinación de la actividad antichagásica de una molécula. ESP P200900242.
69. Leach, A. R., Gillet, V. J., Lewis, R. A., & Taylor, R. Three-dimensional pharmacophore methods in drug discovery. J Med Chem 53(2), 539-558.
70. Hudock, M. P., Sanz-Rodriguez, C. E., Song, Y., Chan, J. M., Zhang, Y., Odeh, S., Kosztowski, T., Leon-Rossell, A., Concepcion, J. L., Yardley, V., Croft, S. L., Urbina, J. A., & Oldfield, E. (2006). Inhibition of Trypanosoma cruzi hexokinase by bisphosphonates. J Med Chem 49(1), 215-223.
71. Barnum, D., Greene, J., Smellie, A., & Sprague, P. (1996). Identification of common functional configurations among molecules. J Chem Inf Comput Sci 36(3), 563-571.
72. Richmond, N. J., Abrams, C. A., Wolohan, P. R., Abrahamian, E., Willett, P., & Clark, R. D. (2006). GALAHAD: 1. pharmacophore identification by hypermolecular alignment of ligands in 3D. J Comput Aided Mol Des 20(9), 567-587.
73. Jones, G., Willett, P., & Glen, R. C. (1995). A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J Comput Aided Mol Des 9(6), 532-549.
74. Cramer, R. D., Patterson, D. E., & Bunce, J. D. (1988). Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 110: 5959–5967.
75. Cramer, R. D., Patterson, D. E., & Bunce, J. D. (1989). Recent advances in comparative molecular field analysis (CoMFA). Prog Clin Biol Res 291: 161-165.
76. Klebe, G., & Abraham, U. (1999). Comparative molecular similarity index analysis (CoMSIA). to study hydrogen-bonding properties and to score combinatorial libraries. J Comput Aided Mol Des 13(1), 1-10.
77. Klebe, G., Abraham, U., & Mietzner, T. (1994). Molecular similarity indices in a comparative analysis (CoMSIA). of drug molecules to correlate and predict their biological activity. J Med Chem 37(24), 4130-4146.
78. Nilsson, J., Wikstrom, H., Smilde, A., Glase, S., Pugsley, T., Cruciani, G., Pastor, M., & Clementi, S. (1997). GRID/GOLPE 3D quantitative structure-activity relationship study on a set of benzamides and naphthamides, with affinity for the dopamine D3 receptor subtype. J Med Chem 40(6), 833-840.
79. El-Sayed, N. M., Myler, P. J., Bartholomeu, D. C., Nilsson, D., Aggarwal, G., Tran, A. N., Ghedin, E., Worthey, E. A., Delcher, A. L., Blandin, G., Westenberger, S. J., Caler, E., Cerqueira, G. C., Branche, C., Haas, B., Anupama, A., Arner, E., Aslund, L., Attipoe, P., Bontempi, E., Bringaud, F., Burton, P., Cadag, E., Campbell, D. A., Carrington, M., Crabtree, J., Darban, H., da Silveira, J. F., de Jong, P., Edwards, K., Englund, P. T., Fazelina, G., Feldblyum, T., Ferella, M., Frasch, A. C., Gull, K., Horn, D., Hou, L., Huang, Y., Kindlund, E., Klingbeil, M., Kluge, S., Koo, H., Lacerda, D., Levin, M. J., Lorenzi, H., Louie, T., Machado, C. R., McCulloch, R., McKenna, A., Mizuno, Y., Mottram, J. C., Nelson, S., Ochaya, S., Osoegawa, K., Pai, G., Parsons, M., Pentony, M., Pettersson, U., Pop, M., Ramirez, J. L., Rinta, J., Robertson, L., Salzberg, S. L., Sanchez, D. O., Seyler, A., Sharma, R., Shetty, J., Simpson, A. J., Sisk, E., Tammi, M. T., Tarleton, R., Teixeira, S., Van Aken, S., Vogt, C., Ward, P. N., Wickstead, B., Wortman, J., White, O., Fraser, C. M., Stuart, K. D., & Andersson, B. (2005). The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309(5733), 409-415.
80. Atwood, J. A., 3rd, Weatherly, D. B., Minning, T. A., Bundy, B., Cavola, C., Opperdoes, F. R., Orlando, R., & Tarleton, R. L. (2005). The Trypanosoma cruzi proteome. Science 309(5733), 473-476.
81. Protein Data Bank. http://www.pdb.org.
82. De Azevedo, W. F., Jr., & Dias, R. (2008). Computational methods for calculation of ligand-binding affinity. Curr Drug Targets 9(12), 1031-1039.
83. Dias, R., & de Azevedo, W. F., Jr. (2008). Molecular docking algorithms. Curr Drug Targets 9(12), 1040-1047.
84. Congreve, M., Chessari, G., Tisi, D., & Woodhead, A. J. (2008). Recent developments in fragment-based drug discovery. J Med Chem 51(13), 3661-3680.
85. Huang, S.Y., Grinter, S.Z. Zou, X. (2011). Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future diretions. Phys. Chem. Chem. Phy. 12, 12899-12908.
86. Zhong, S., Zhang, Y., Xin, Z. (2011) Rescoring ligand docking poses. Curr. Opin. Drug. Discov. Devel. 13, 326-334.
87. Kuntz, I. D., Blaney, J. M., Oatley, S. J., Langridge, R., & Ferrin, T. E. (1982). A geometric approach to macromolecule-ligand interactions. J Mol Biol 161(2), 269-288.
88. Perola, E., Xu, K., Kollmeyer, T. M., Kaufmann, S. H., Prendergast, F. G., & Pang, Y. P. (2000). Successful virtual screening of a chemical database for farnesyltransferase inhibitor leads. J Med Chem 43(3), 401-408.
89. Venkatachalam, C. M., Jiang, X., Oldfield, T., & Waldman, M. (2003). LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model 21(4), 289-307.
90. Rarey, M., Kramer, B., Lengauer, T., & Klebe, G. (1996). A fast flexible docking method using an incremental construction algorithm. J Mol Biol 261(3), 470-489.
91. Jones, G., Willett, P., Glen, R. C., Leach, A. R., & Taylor, R. (1997). Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267(3), 727-748.
92. Morris, G. M. G., D.S.; Halliday, R.S., Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry 19(14).
93. Kontoyianni, M., Madhav, P., Suchanek, E., & Seibel, W. (2008). Theoretical and practical considerations in virtual screening: a beaten field? Curr Med Chem 15(2), 107-116.
94. ZINC. http://blaster.docking.org/zinc.
95. Canyuk, B., Medrano, F. J., Wenck, M. A., Focia, P. J., Eakin, A. E., & Craig, S. P., 3rd (2004). Interactions at the dimer interface influence the relative efficiencies for purine nucleotide synthesis and pyrophosphorolysis in a phosphoribosyltransferase. J Mol Biol 335(4), 905-921.
96. Canyuk, B., Focia, P. J., & Eakin, A. E. (2001). The role for an invariant aspartic acid in hypoxanthine phosphoribosyltransferases is examined using saturation mutagenesis, functional analysis, and X-ray crystallography. Biochemistry 40(9), 2754-2765.
97. Focia, P. J., Craig, S. P., 3rd, Nieves-Alicea, R., Fletterick, R. J., & Eakin, A. E. (1998). A 1.4 A crystal structure for the hypoxanthine phosphoribosyltransferase of Trypanosoma cruzi. Biochemistry 37(43), 15066-15075.
98. Freymann, D. M., Wenck, M. A., Engel, J. C., Feng, J., Focia, P. J., Eakin, A. E., & Craig, S. P. (2000). Efficient identification of inhibitors targeting the closed active site conformation of the HPRT from Trypanosoma cruzi. Chem Biol 7(12), 957-968.
99. Ferreira, R. S., Simeonov, A., Jadhav, A., Eidam, O., Mott, B. T., Keiser, M. J., McKerrow, J. H., Maloney, D. J., Irwin, J. J., & Shoichet, B. K. (2010). Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J Med Chem 53(13), 4891-4905.