ARTÍCULO |
Síntesis de Inhibidores Selectivos de MMP-2 utilizando Química Click
José María Zapico1, Pilar Serra1, Josune García Sanmartín2, Kamila Filipiak1, 3, Alfredo Martínez2, Sonsoles Martín Santamaría1, Beatriz de Pascual-Teresa1, Ana Ramos1*
1Departamento de Química, Facultad de Farmacia, Universidad San Pablo CEU, Urbanización Montepríncipe, 28668 Boadilla del Monte, Madrid, Spain. 2Centro de Investigación Biomédica de La Rioja (CIBIR) C/ Piqueras 98, 26006 Logroño, Spain.3Department of Molecular Biology, Faculty of Mathematics and Natural Sciences, The John Paul II Catholic University of Lublin, 20-718 Lublin, Poland.
Recibido el 4 de abril de 2011.
e-mail: bpaster@ceu.es
RESUMEN
Siguiendo una estrategia de diseño basado en fragmentos, se describe la síntesis de una nueva serie de inhibidores de MMP-2. Para ello, se parte de un fragmento que contiene simultáneamente un grupo hidroxamato como Zinc Binding Group (ZBG) y un grupo azida. Esta subunidad se conecta mediante química “click" con otros fragmentos lipófilos que contienen un alquino terminal y que han sido seleccionados para interaccionar de manera selectiva con el subsitio S1’ de la MMP-2. Los compuestos sintetizados más activos, 20 y 21, presentan una alta potencia inhibitoria en MMP-2. Además, el compuesto 20 presenta un prometedor perfil de selectividad frente a algunas metaloproteasas consideradas anti-diana en cáncer, como MMP-8 y MMP-9. |
Palabras clave: Inhibidores MMP-2; Hidroxamatos; Química “click”; Modelado molecular; Evaluación biológica.
abstract
Synthesis of Selective MMP-2 Inhibitors Using Click Chemistry
A new series of selective MMP-2 inhibitors is described, following a fragment-based drug design approach. A fragment containing an azide group and a well known hydroxamate ZBG, was synthesized. A click chemistry reaction was used to connect the azide to lipophilic alkynes selected to interact selectively with the S1’ subunit of MMP-2. The most active compounds, 20 and 21, displayed high values of IC50 against MMP-2. In addition, compound 20 has shown also a promissing selectivity profile against some antitarget metalloproteinases in cancer, such as MMP-8, and MMP-9. |
Key words: MMP-2 inhibitors; Hydroxamates; Click chemistry; Docking; Biological evaluation.
1. introducCIÓn
Las Metaloproteasas de la Matriz (MMPs), también conocidas como matrixinas, son una familia de enzimas estructuralmente relacionadas que poseen zinc en su centro catalítico. Las MMPs son responsables del remodelado y la degradación de la matriz extracelular, por lo que están implicadas en una gran variedad de procesos biológicos, tales como el desarrollo embrionario, enfermedades neurológicas (1, 2), artritis (3), enfermedades cardiovasculares (4, 5), y varios procesos relacionados con el cáncer, como la angiogénesis, apoptosis, proliferación celular y metástasis (6-8).
En la actualidad, se conocen en vertebrados al menos 26 MMPs (9), 23 de las cuales han sido detectadas en humanos (10). Están clasificadas en seis grupos: colagenasas: MMP1, MMP8, MMP13 y MMP18; gelatinasas: MMP2 y MMP9; estromelisinas: MMP3, MMP10 y MMP11; matrilisinas: MMP7 y MMP26; MMPs de la membrana: MMP14 a MMP17, MMP24 y MMP25 y otras MMPs (10).
Originalmente, se pensó que las MMPs estaban implicadas fundamentalmente en la invasión y metástasis, debido al remodelado que provocan en la matriz extracelular, permitiendo de ese modo a las células tumorales acceder a los vasos sanguíneos y linfáticos. Este mecanismo se propuso debido al incremento de la capacidad de invasión que poseen las líneas celulares que sobreexpresan MMPs. Más recientemente, se ha visto que las MMPs pueden estar implicadas también en el crecimiento de tumores primarios. La remodelación de la matriz extracelular en las proximidades de un tumor primario puede proporcionarle los requerimientos especiales necesarios para su crecimiento (11).
Se ha descrito un gran número de compuestos que actúan como inhibidores de MMPs y que han permitido deducir cuáles son las características estructurales necesarias para presentar actividad y que se pueden resumir en:
-Un grupo funcional conocido como Zinc-Binding Group, ZBG (ácido hidroxámico, ácido carboxílico, sulfhidrilo, etc.), capaz de unirse al Zn2+ catalítico.
-Al menos un grupo funcional capaz de formar un enlace de hidrógeno con el esqueleto de la proteína.
-Una o más cadenas laterales que puedan establecer interacciones de van der Waals con los subsitios de la enzima.
Los hidroxamatos han sido los ZBGs más utilizados en el desarrollo de inhibidores de MMPs, ya que se coordinan al zinc formando un quelato a través de dos enlaces. La capacidad de unión de estos inhibidores no se debe únicamente a esta coordinación con el Zn2+ catalítico, sino que la afinidad y, sobre todo, la selectividad entre las diferentes MMPs, se debe fundamentalmente a la interacción de otros grupos de la molécula con diferentes subsitios de la enzima. Especialmente interesante, es la región conocida como sitio S1’, por ser una de las que más diferencias presenta entre las diferentes MMPs (12, 13). Modificaciones realizadas sobre la parte del inhibidor que interacciona con este subsitio (P1’) han llevado a un aumento (14, 15) o disminución de la selectividad (16, 17).
Por el momento, solamente se han validado experimentalmente como dianas contra el cáncer las MMPs 1, 2 y 7 (9, 18). De ellas, la MMP-2 se está erigiendo en los últimos años en la más interesante para el diseño de agentes antitumorales. La MMP-1 parece ser la causante del síndrome musculo esquelético observado clínicamente con inhibidores poco selectivos de MMPs (19). Por otro lado, la inhibición de otras MMPs como la 3 y 8 puede incrementar la tumorigénesis y metástasis (20, 21). La MMP-9 es una enzima muy particular, ya que su inhibición puede ser útil en el tratamiento de pacientes con cáncer en una etapa inicial, sin embargo debe evitarse en pacientes en etapas avanzadas de la enfermedad (9).
El papel de la MMP-2 en cáncer está relacionado con la estimulación del crecimiento tumoral, angiogénesis y metástasis, a través de su implicación en la degradación de la matriz extracelular (22). Además, se ha visto que en muestras de tumor humano está sobreexpresada y se ha identificado en asociación con células altamente invasivas. Por estas razones se ha considerado como una importante diana contra el cáncer, y el diseño de inhibidores selectivos de esta enzima puede conducir a interesantes agentes anticancerosos (23).
El objetivo de nuestro trabajo, es la búsqueda de inhibidores selectivos de MMP-2 y especialmente frente a MMP-9, otra gelatinasa con una gran homología de secuencia en el sitio activo y cuya inhibición, como ya hemos mencionado, nos interesa evitar.
Entre los inhibidores de MMP-2 descritos en la bibliografía, cabe destacar los hidroxamatos de estructura general 1 (Figura 1), que poseen un ciclo de tetrahidropirano y una sulfona en posición a respecto al grupo hidroxamato (24-27). Algunos de ellos, presentan una elevada actividad en MMP-2, MMP-9 y MMP-13, mientras que son inactivos frente a MMP-1.
Por esta razón, hemos empleado el esqueleto de a-sulfona-a-tetrahidopiranil hidroxamato en el diseño de nuevos inhibidores que, manteniendo la potente actividad frente a MMP-2 y selectividad frente a MMP-1, sean también selectivos frente a otras metaloproteasas, especialmente MMP-9.
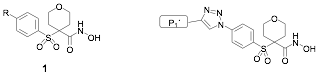
Figura 1.- Inhibidores de MMP-2 descritos con estructura general 1 (izquierda) y estructura general de los inhibidores descritos en este trabajo.
Para ello, hemos utilizado una estrategia basada en el uso de química “click” para conectar el fragmento que contiene el ZBG con otros fragmentos de carácter lipófilo, seleccionados para interaccionar con el subsitio S1´de la enzima, de acuerdo a los datos recogidos en la bibliografía sobre inhibidores de MMP-2 (28).
La química “click” y, concretamente, la reacción de cicloadición 1,3-dipolar entre una azida y un alquino catalizada por cobre (I) (Copper Azide Alkyne Cycloaddition, CuAAC) se ha utilizado en el descubrimiento de inhibidores de diferentes enzimas, como por ejemplo acetilcolinesterasa (29-31), proteasa del HIV-1 (32, 33), tirosina quinasa de Abelson (34), trans-sialidasa de tripanosoma cruzi(35), y caspasas (36). Se ha empleado también esta estrategia en la búsqueda de inhibidores selectivos de MMP-7 frente a termolisina y colagenasa (37).
2. MATERIAL Y MÉTODOS
2.1. Síntesis general
Las reacciones sensibles al aire se realizaron bajo atmósfera de argón. Los disolventes utilizados se purificaron por destilación antes de su uso: el tetrahidrofurano sobre sodio-benzofenona bajo atmósfera de argón y el acetonitrilo sobre hidruro cálcico.
La separación de los crudos de reacción y la purificación de los compuestos obtenidos se llevó a cabo por cromatografía en columna utilizando gel de sílice Merck 230-240 mesh y el eluyente indicado en cada caso. El análisis de los productos de reacción se realizó por cromatografía en capa fina (kiesegel 60F-254). Para la detección de los compuestos se utilizó luz ultravioleta (λ = 254 y 365 nm) y PMA al 5% en etanol.
Los puntos de fusión se determinaron en un tubo capilar abierto en un aparato Stvert Scientific SMP3 y están sin corregir. Los compuestos se caracterizaron por 1H RMN, 13C RMN, espectrometría de masas y análisis elemental.
2.2. Síntesis del fragmento F1 (azida2)
[(4-Nitrofenil)sulfanil]acetato de metilo (3). A una disolución de 4-nitrobencenotiol (5,05 g, 26,01 mmol) en DMF (50 mL) y a 0 ºC se añade K2CO3 (5,39 g, 39,02 mmol). La mezcla de reacción se agita 15 min a 0 ºC, a continuación se añade bromoacetato de metilo (2,65 mL, 28,62 mmol) y se agita a temperatura ambiente toda la noche. Posteriormente, se diluye con AcOEt (200 mL) y la disolución se lava sucesivamente con una disolución acuosa saturada de NH4Cl y salmuera. La fase orgánica se seca (MgSO4), filtra y evapora a sequedad. El residuo obtenido se purifica por cromatografía en columna sobre gel de sílice, empleando como eluyente hexano: AcOEt 85:15 para dar 3 (5,80 g, 98%) como un sólido blanco, P.F. 69,8-71.2 ºC (EtOH).
[(4-Nitrofenil)sulfonil]acetato de metilo (4). A una disolución de 3 (5,72 g, 25,17 mmol) en MeOH (50 mL) y agua (5 mL), se añade a 0 ºC Oxone (38,69 g, 62,93 mmol). La reacción se agita a temperatura ambiente durante 2 h. Los restos de Oxone se eliminan por filtración y se lavan con MeOH. La fracción metanólica se concentra a vacío, se disuelve en AcOEt y se lava con una disolución acuosa saturada de NaHCO3 y salmuera. La fase orgánica se seca (MgSO4), se filtra, se evapora a sequedad y se recristaliza en AcOEt para dar 4 (5,84 g, 90%) como un sólido amarillento, P.F. 130,3-131,8 ºC (AcOEt).
4-[(4-Nitrofenil)sulfonil]tetrahidro-2H-pirano-4-carboxilato de metilo (5). A una disolución de 4 (3,00 g, 11,57 mmol) en DMF (30 mL) a 0 ºC se añade K2CO3 (4,00 g, 28,93 mmol). La mezcla de reacción se agita a temperatura ambiente durante 15 min y a continuación se añaden bis-(2-bromoetil)eter (2,95 g, 12,73 mmol), DMAP (85 mg, 0,69 mmol) y yoduro de tetrabutilamonio (256 mg, 0,69 mmol). La mezcla de reacción se agita a temperatura ambiente 24 h y a continuación se vierte sobre HCl 1N (100 mL). El sólido obtenido se filtra y se lava con hexano para dar, tras recristalización en AcOEt, 5 como un sólido amarillento (4,66 g, 76%), P.F. 192,8-194,6 ºC.
Ácido 4-[(4-nitrofenil)sulfonil]tetrahidro-2H-pirano-4-carboxílico (6). A una disolución de 5 (3,94 g, 11,96 mmol) en THF (40 mL) se añade una disolución de NaOH 1N (120 mL) y la mezcla se agita 4 h a temperatura ambiente. A continuación se concentra, se suspende en H2O y se lava con AcOEt. La fase acuosa se acidifica a pH 2 con HCl 3N y se extrae con AcOEt. La fase orgánica se seca (MgSO4), se filtra y evapora a sequedad para dar 6 (3,40 g, 90%) como un sólido blanco, P.F. 239,3-240,3 ºC (descompone).
Ácido 4-[(4-aminofenil)sulfonil]tetrahidro-2H-pirano-4-carboxílico (7). A una disolución de 6 (3,94 g, 12,50 mmol) en EtOH (150 mL) y H2O (50 mL) se añade Pd/C (10%) (250 mg) y se introduce en un sistema de hidrogenación Parr, manteniendo una presión de hidrógeno de 60 p.s.i durante 5 h a temperatura ambiente. Se elimina el paladio por filtración y a continuación el disolvente a vacío para obtener 7 (3,40 g, 95%) como un sólido blanco, P.F. 227,6-228,8 ºC.
Ácido 4-[(4-azidofenil)sulfonil]tetrahidro-2H-pirano-4-carboxílico (8). El compuesto 7 (1,99 g, 6,98 mmol) se disuelve en CH3CN anhidro (50 mL) y bajo argon y a 0 ºC se añaden ButONO (1,24 mL, 10,47 mmol) y TMSN3 (1,10 mL, 8,38 mmol) gota a gota. La mezcla de reacción se agita a 0 ºC durante 30 min y posteriormente 3 h a temperatura ambiente. A continuación, se evapora a sequedad y el residuo se purifica por cromatografía en columna sobre gel de sílice, empleando como eluyente CH2Cl2:MeOH 97,5:2,5 para obtener 8 (2,04 g, 94%) como un sólido amarillento, P.F. 191,4-192,4 ºC (descompone).
4-[(4-Azidofenil)sulfonil]-N-(tetrahidro-2H-piran-2-iloxi)tetrahidro-2H-pirano-4-carboxamida (9). A una disolución de 8 (1,65 g, 5,30 mmol) en DMF (20 mL) se añaden HOBT (859 mg, 6,36 mmol), NMM (1,75 mL, 15,90 mmol), NH2-OTHP (1,24 g, 10,60 mmol) y EDCI (1,42 g, 7,42 mmol). La reacción se deja agitando toda la noche a temperatura ambiente y a continuación se diluye con AcOEt (60 mL) y se lava sucesivamente con una disolución acuosa saturada de NH4Cl y salmuera. La fase orgánica se seca (MgSO4), filtra y evapora a sequedad. El sólido obtenido se recristaliza en EtOH y agua para obtener 9 (1,97 g, 91%) como un sólido anaranjado, P.F. 194,2-195,8 ºC.
4-[(4-Azidofenil)sulfonil]-N-hidroxitetrahidro-2H-pirano-4-carboxamida (2). A una disolución de 9 (1,92 g, 4,69 mmol) en dioxano (10 mL) se añaden HCl 4N en dioxano (5,86 mL, 23,43 mmol) y MeOH (10 mL). La reacción se agita 2 h a temperatura ambiente y a continuación se concentra a vacío. El sólido obtenido se recristaliza de EtOH y agua para dar 2 (1,36 g, 89%) como un sólido naranja, P.F. 197,2-198,1 ºC (descompone).
2.3. Síntesis del fragmento F2 (alquinos)
N-(Prop-2-in-1-ilcarbamoil)bencenosulfonamida (10). A una disolución de propargilamina (1,48 g, 26,38 mmol) en CH3CN anhidro (40 mL) se añade, bajo argón, isocianato de bencenosulfonilo (5,09 g, 26,38 mmol). La mezcla de reacción se agita toda la noche a temperatura ambiente. A continuación, se concentra a vacío y el sólido obtenido se recristaliza de AcOEt para dar 10 (5,47 g, 87%) como un sólido blanco, P.F. 161,8-163,6 ºC.
[3-(4-Pentilfenil)prop-1-inil] trimetilsilano (13) A una mezcla de virutas de magnesio (0,34 g, 14,04 mmol) y yodo (10 mg) en THF anhidro (2 mL), se añade en atmósfera de argón una fracción (1 mL) de una disolución de 1-bromo-4-pentilbenceno (3,00 g, 12,81 mmol) en THF anhidro (10 mL). Una vez empezada la formación del magnesiano y tras la posterior adición de 10 mL de THF anhidro, se completa la adición, gota a gota, de la disolución del bromuro de arilo. La mezcla de reacción se calienta a reflujo 2 h y, una vez fría, el exceso de magnesio se elimina por filtración vía cánula. A la disolución restante se le añade (3-bromoprop-1-inil)trimetilsilano (2,0 g, 10,25 mmol) y la mezcla se calienta a reflujo 12 h, se enfría y se concentra a vacío. Se añade una disolución de HCl 1N (50 mL) al crudo y se extrae con Et2O. La fase orgánica se seca (MgSO4), filtra y evapora a sequedad. El residuo obtenido se purifica por cromatografía en columna sobre gel de sílice, empleando como eluyente hexano, para obtener (13) (708 mg, 21%) como un líquido incoloro.
[3-(4-Fenoxifenil)prop-1-inil]trimetilsilano (14) Siguiendo el método anterior se parte de 1-bromo-4-fenoxibenceno (4,00 g, 16,06 mmol), virutas de magnesio (0,42 g, 17,29 mmol), yodo (10 mg) y (3-bromoprop-1-inil)trimetilsilano (2,36 g, 12,35 mmol). Tras la purificación cromatográfica con hexano:CH2Cl2 (95:5) como eluyente se obtiene 14 (1,15 g, 33%) como un líquido incoloro.
1-Pentil-4-(2-propin-1-il) benceno (11) El alquino 13 (618 mg, 2,39 mmol) se disuelve en EtOH (10 mL) y se le añaden gota a gota una disolución de nitrato de plata (609 mg, 3,59 mmol) en agua (3 mL) y EtOH (7 mL). La mezcla de reacción se agita a temperatura ambiente 30 min y a continuación se añade una disolución de KCN (1,58 g, 23,91 mmol) en agua (3 mL). La mezcla se extrae con Et2O, y la fase orgánica se lava con agua y salmuera, se seca (MgSO4), filtra y evapora a sequedad. Se obtiene un residuo que se purifica por cromatografía en columna sobre gel de sílice, empleando hexano como eluyente, para obtener 11 (430 mg, 97%) como un aceite incoloro que descompone con el tiempo.
1-Fenoxi-4-(2-propin-1-il)benceno (12) Siguiendo el método anterior, se parte de 14 (1,15 g, 4,10 mmol), AgNO3 (1,05 g, 6,15 mmol) y KCN (2,67 g, 41,01 mmol). Tras la purificación cromatográfica empleando como eluyente hexano:CH2Cl2 9:1 se obtiene 12 (734 mg, 86%) como un aceite incoloro que descompone con el tiempo.
2.4. Síntesis general de triazoles
A una mezcla de la azida 2 (1 equiv) y alquino (1,2-1,3 equiv.) en ButOH y H2O (V/V = 1:1, 5 mL) y bajo argón, se añade ascorbato sódico (2 equiv. de una disolución acuosa 1 M recientemente preparada) y sulfato de cobre (II) pentahidratado (0,5 equiv. de una disolución acuosa 0,25 M). La mezcla de reacción se agita vigorosamente toda la noche, tras lo que se añade agua (20 mL) y hielo. El precipitado obtenido se filtra y se lava con agua (2 x 10 mL) y hexano (2 x 10 mL). El sólido obtenido se disuelve en una mezcla de CH2Cl2:MeOH:NH3 (acuoso) 6:3:1, se filtra sobre gel de sílice y se concentra a vacío para obtener los compuestos deseados (15-25).
2.5. Estudios de modelado molecular
Estudios de docking: GLIDE (grid-based ligand docking with energetics) (38).
La diana utilizada para los estudios de docking, ha sido la estructura resuelta por RMN del dominio catalítico de la MMP2 con un inhibidor de tipo hidroxamato, el compuesto i52 (1hov.pdb) (39). Para estos estudios, se ha utilizado el modelo 1 de la estructura depositada en el pdb, de acuerdo con los resultados obtenidos en estudios previos con esta misma diana (40, 41). Esta estructura se optimiza y minimiza usando la aplicación Protein Preparation Wizard intregrada en el paquete de GLIDE. Seguidamente, se crean los mapas de interacción del sitio activo de la MMP-2 usando la aplicación Receptor Grid Generation y se selecciona al ligando i52 como centro de la caja. Todos los compuestos a estudiar se preparan usando la aplicación Ligprep. El campo de fuerzas OPLS-2005 se selecciona para la preparación de ambos, proteína y ligandos. Los estudios de docking se establecen para llevarse a cabo de forma aleatoria.
Para cada estudio se selecciona que el programa busque 10.000 conformaciones por pose (o solución de docking) y un número máximo de 10 poses por compuesto. El método seleccionado para el estudio es el XP (Extra-Precision) docking (42).
2.6. Zimografía
Como fuente de gelatinasas se emplea suero de sangre humana perteneciente a voluntarios sanos. En cada pocillo del gel para zimografía, con un 10% de gelatina, se añade la misma cantidad de suero (1,5 ml) (Invitrogen, Carlsbad, CA,USA). Tras la electroforesis, el gel se incuba durante 30 min en un tampón renaturalizante y otros 30 min en un tampón de revelado, ambos a temperatura ambiente. A continuación, los geles se cortan en líneas individuales y se incuban en presencia de los inhibidores en el tampón de revelado toda la noche a 37 ºC. Tras varios lavados los fragmentos del gel se revelan con Simply Blue (Invitrogen), se elimina el revelador y se escanean con Odyssey (Li-cor, Lincoln, Nebraska, USA). La actividad sobre la gelatina de cada enzima (MMP2, 72 kDa; MMP9, 92 kDa) se cuantifica por análisis de imagen (Image-J, NIH, Bethesda, MD, USA) y los valores de CI50 se calculan con MS Excel en XLfit (IDBS, UL, Vs. 5.0).
2.7. Ensayo de inhibición en MMPs
Las medidas de actividad en las diferentes MMPs se lleva a cabo usando el ensayo colorimétrico comercial MMP Inhibitor Profiling Kit (Enzo Life Science International, Inc.). La absorbancia se mide a 414 nm (microplate photometer Thermo Scientific Multiscan FC). La reacción enzimática se lleva a cabo a 37 ºC. Las correspondientes MMPs, se incuban por triplicado con al menos seis concentraciones de cada inhibidor. Tras la adición del sustrato, el incremento de absorbancia se recoge cada minuto durante 20 min. Se representa gráficamente la densidad óptica frente al tiempo para obtener la ecuación de velocidad. El porcentaje de actividad residual para cada compuesto se calcula usando la siguiente fórmula: % de actividad remanente = velocidad en presencia del inhibidor / velocidad del control x 100. Se emplea como control positivo el inhibidor NNGH (43).
3. RESULTADOS Y DISCUSIÓN
En el esquema 1 se muestra la reacción “click" utilizada en las síntesis de los inhibidores que se describen en este trabajo. Se parte del fragmento F1, que contiene simultáneamente el ZBG (hidroxamato) y el grupo funcional azida, y de diferentes fragmentos F2, que contienen un alquino terminal de naturaleza generalmente hidrófoba para interaccionar con el subsitio S1’ de la enzima. Ambos fragmentos se han unido utilizando la CuAAC, dando lugar a los correspondientes 1,2,3-triazoles 1,4-disustituidos.
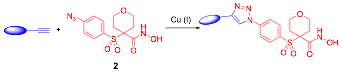
Esquema 1.- Método general de síntesis de los 1,2,3-triazoles 1,4-disustituidos.
El fragmento F1 que contiene el ZBG posee además un grupo sulfona. Este grupo es muy importante por su capacidad de establecer un enlace de hidrógeno con el esqueleto peptídico de la enzima, dirigiendo el sustituyente hidrófobo hacia el bolsillo S1’. Los alquinos utilizados se han seleccionado en un trabajo previo en el que se ha realizado una búsqueda bibliográfica de inhibidores de MMP-2, cuyas interacciones con la enzima se analizaron mediante técnicas de modelado molecular (44). Algunos de los alquinos son comerciales y otros son fácilmente asequibles mediante la síntesis que se detalla más adelante.
La síntesis de la azida 2 se muestra en el esquema 2. El primer paso consiste en la alquilación del 4-nitrobencenotiol, que conduce al sulfuro 3 con un rendimiento excelente (98%). A continuación, se obtiene la sulfona 4 por oxidación con oxone® y se forma el tetrahidropirano 5 por dialquilación en la posición a de la sulfona. La saponificación del éster metílico con hidróxido sódico para dar 6 y la posterior hidrogenación catalítica del grupo nitro conduce al aminoácido 7, que se convierte en el producto final tras las etapas de transformación del grupo amino en azida para dar 8, acoplamiento con o-THP hidroxilamina formando 9 y posterior desprotección del grupo THP para dar 2. El rendimiento global del proceso es del 46% para los ocho pasos de reacción.
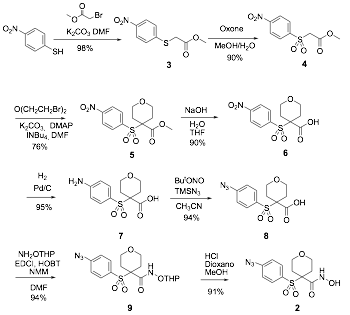
Esquema 2.- Síntesis de la azida 2.
La mayoría de los alquinos elegidos son comerciales (Figura 2). Sin embargo ha sido necesaria la síntesis de 10-12 (esquema 3).
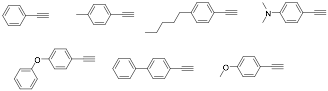
Figura 2.- Alquinos disponibles comercialmente.
El alquino 10 se obtiene fácilmente y con buen rendimiento a partir de propargilamina e isocianato de bencenosulfonilo. Para la síntesis de 11 y 12, en primer lugar se sintetiza el magnesiano del correspondiente bromuro de arilo, que se hace reaccionar con bromuro de propargilo protegido con el grupo trimetilsililo, obteniendose los alquinos 13 y 14. En un segundo paso, se elimina el grupo protector para dejar libre el alquino terminal.
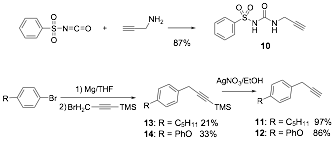
Esquema 3.- Síntesis de los alquinos 10-12.
Una vez sintetizada la azida 2 y los alquinos, se procedió a su acoplamiento mediante la reacción de cicloadición 1,3-dipolar catalizada por cobre para obtener los triazoles 15-25 con rendimientos moderados (27-79%) (esquema 4).
Se utilizó el sistema cobre (II)/ascorbato sódico como catalizador, ensayándose una gran variedad de condiciones para optimizar la reacción. El empleo de bajas cantidades de CuSO4 (0,25-2 mol%) en ButOH/H2O y ácido ascórbico (5-10 mol %) condujo a una baja conversión de los productos de partida y no se observó progreso en la reacción, incluso después de 3 días o bajo calentamiento moderado.
Como consecuencia, la cantidad de catalizador se incrementó progresivamente, obteniéndose los mejores resultados tras la adición de 0,5 equivalentes de CuSO4 y dos equivalentes de ácido ascórbico y en atmósfera inerte, ya que en presencia de oxígeno fue necesario el calentamiento de la mezcla de reacción a 80 ºC para una conversión total. Como disolvente para la reacción se empleó una mezcla 1:1 de H2O:ButOH, ya que otros disolventes como N,N-dimetilformamida no condujeron a mejores resultados.
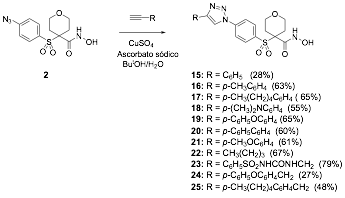
Esquema 4.- Síntesis de los triazoles 15-25.
Evaluación Biológica de los triazoles
A continuación, se empleó la técnica de zimografía para la evaluación de la inhibición de la actividad enzimática de MMP-2 y MMP-9 de todos los triazoles 15-25. Los resultados se muestran en la Tabla 1.
Hay que destacar que todos los compuestos presentan una actividad inhibitoria muy alta frente a MMP-2, con valores de CI50 del orden picomolar para los compuestos 15-21 y superior a la obtenida para el BiPS en este mismo ensayo (inhibidor comercial de MMP-2 y MMP-9).
Por otro lado, los compuestos 23, 24 y 25 presentan peores valores de inhibición que el resto de triazoles, con valores de CI50 en el rango de nanomolar.
Tabla 1. Actividad inhibitoria frente a MMP-2 y MMP-9 para los tetrahidropiranos 15-25.
Compuesto |
R |
MMP-2 CI50, nM |
MMP-9 CI50, nM |
Selectividad MMP9/MMP2 |
15 |
|
1.034´10-2 |
3.06´10-2 |
2.96 |
16 |
|
2.45´10-3 |
11.11´10-3 |
4.53 |
17 |
|
7.75´10-3 |
13.57´10-3 |
1.75 |
18 |
|
1.91´10-3 |
14.63´10-3 |
7.65 |
19 |
|
10.02´10-3 |
32.69´10-3 |
3.26 |
20 |
|
1.94´10-3 |
18.83´10-3 |
9.69 |
21 |
|
9.47´10-3 |
51.47´10-3 |
5.43 |
22 |
|
53.25´10-3 |
127.54´10-3 |
2.40 |
23 |
|
1.05 |
> 50.00 |
- |
24 |
|
1.18 |
> 50.00 |
- |
25 |
|
6.79 |
-* |
- |
BiPS |
22.28´10-3 |
53.40´10-3 |
2.40 |
*No fue posible calcular un valor específico de CI50 para la MMP-9. Sin embargo, el valor de inhibición ha sido menor que el obtenido para MMP-2.
El valor específico de CI50 en MMP-2 para todos los compuestos ha sido más bajo que los valores para MMP-9, presentando en algunos casos una relación de inhibición MMP-2/MMP-9 muy interesante. Por ello, se seleccionaron 20 y 21 para un estudio posterior del perfil de inhibición en una batería de MMPs.
Estudios de modelado molecular de los triazoles
Para explicar los resultados de actividad observados y tratar de elucidar el modo de unión de estos compuestos a MMP-2, se ha llevado a cabo un estudio de modelado molecular mediante técnicas de docking. Todos los compuestos con una actividad del orden picomolar (15-22) tienen, según los experimentos de docking, un comportamiento similar a i52 (Figura 3) (inhibidor de MMP-2 presente en la estructura de RMN del PDB 1hov).
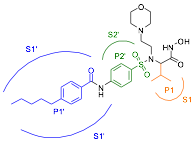
Figura 3.- Estructura química de i52 donde se muestran las interacciones con los principales subsitios del centro activo de MMP-2.
En la Figura 4 (amarillo) se representa el modo de unión del compuesto 20, que es uno de los más activos de la serie (CI50 = 1,94 pM) y selectivo frente a MMP-9. Analizando los datos obtenidos, se observa que los dos átomos de oxígeno del grupo funcional hidroxamato se coordinan con el zinc catalítico adoptando, junto con las histidinas, una disposición de bipirámide trigonal distorsionada. Además, la cadena lateral se dirige hacia el subsitio S1’ de la MMP-2, estableciendo interacciones hidrófobas en dicho bolsillo. Se destacan las interacciones con los aminoácidos Phe148, Phe115, Leu150 y Thr145, ya que son los más implicados en la selectividad. La Thr145 está presente exclusivamente en gelatinasas (MMP-2 y MMP-9), mientras que la Phe148 distingue entre ambas, ya que solo se encuentra presente en el sitio activo de la MMP-2, por lo que las interacciones con estos aminoácidos pueden explicar la selectividad observada.
En el caso del compuesto 15 (verde, Figura 4), cuya actividad baja en un orden de 10 unidades en el ensayo de zimografía, los diferentes resultados del estudio del modo de unión no muestran una clara interacción entre el grupo hidroxamato y el átomo de zinc catalítico. Ello puede deberse a que la cadena lateral más corta, permite un acceso profundo del compuesto dentro del bolsillo hidrófobo S1’ que está favorecido por la formación de un enlace de hidrógeno entre el grupo hidroxamato y el grupo NH de la Leu83 (Figura 4). Según este modo de unión, parece que es uno de los oxígenos del grupo sulfona el que establece una coordinación con el átomo de zinc. En la Figura 4 se representan los resultados de docking del compuesto 15 (verde) y se comparan con los resultados obtenidos para 20 (amarillo).
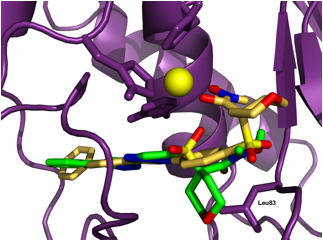
Figura 4.- Modelo del modo de unión obtenido para los compuestos 15 (verde) y 20 (amarillo).
La diferencia más significativa en los valores de actividad se encuentra al pasar a los compuestos 24 y 25, que son análogos de 19 y 17, respectivamente, en los que se ha introducido un grupo metileno adicional entre el fenilo y el triazol. Los resultados de actividad en zimografía mostraron que esa pequeña variación de la estructura conduce a un descenso acusado de la inhibición frente a MMP-2 (1000 unidades de actividad), y MMP-9 (CI50 > 50 nM). En el caso de 24 los estudios de docking muestran que en ninguna de las soluciones de menor energía el sustituyente p-fenoxifenilo se sitúa en el bolsillo S1’, mientras que para 25, aquellas soluciones que colocan la cadena de p-pentilfenilo en el bolsillo S1’, no permiten una coordinación eficaz con el átomo de zinc.
Estudios de actividad en diferentes MMPs
Los compuestos 20 y 21 se seleccionaron para llevar a cabo estudios más amplios de selectividad. Este experimento se realizó sobre 10 MMPs diferentes, utilizando un ensayo colorimétrico comercial de la compañía Biomol, que permite la medida cuantitativa de la inhibición de las diferentes MMPs como un porcentaje de la actividad remanente de cada una de ellas.
En la Tabla 2 se recogen los valores de las actividades inhibitorias de estos dos compuestos.
El compuesto 20 muestra un valor de CI50 = 1,4 nM para MMP-2 en estas condiciones. Sin embargo, es aún más interesante la alta selectividad que presenta en su interacción frente a otras MMPs. Así, su actividad frente a MMP-1 (CI50>1000) y MMP-7 (CI50>100), dos metaloproteasas que poseen un subsitio S1’ poco profundo es muy inferior (45). Además, es también selectivo frente a otras MMPs caracterizadas por poseer, al igual que la MMP-2, un bolsillo S1’ profundo como son la MMP-8 (CI50>100 nM), MMP-14 (CI50 = 65 nM) y MMP-9 (CI50 = 98 nM). La presencia del grupo bifenilo, que confiere una gran rigidez y adecuada hidrofobicidad y longitud para introducirse e interaccionar perfectamente en el subsitio S1’, debe ser la responsable de la gran actividad y selectividad frente a otras MMPs. En cuanto al compuesto 21, en el que el sistema de bifenilo rígido se ha sustituido por un grupo p-metoxifenilo más flexible, presenta una potencia ligeramente mayor en MMP-2 (0,3 nM). Sin embargo, su patrón de selectividades frente a otras MMPs es algo mas reducida.
Tabla 2. Actividad inhibitoria de 20 y 21 en diferentes MMPs.
20 CI50, nM |
21 CI50, nM |
|
MMP-1 |
>1000 |
>1000 |
MMP-2 |
1.4 |
0.3 |
MMP-3 |
17.2 |
9.6 |
MMP-7 |
>100 |
70 |
MMP-8 |
>100 |
6.1 |
MMP-9 |
98 |
11.3 |
MMP-10 |
51 |
7.8 |
MMP-12 |
3.2 |
1.2 |
MMP-13 |
0.9 |
1.4 |
MMP14 |
65 |
8.2 |
En resumen, este estudio nos ha permitido describir un nuevo inhibidor de MMP-2 (20) con una gran potencia (nanomolar en el ensayo colorimétrico y picomolar en zimografía) y una interesante selectividad frente a MMP-9. Además, es inactivo frente a MMP-1, MMP-7 y MMP-8 y presenta una actividad considerablemente menor frente a MMP-3, MMP-4 y MMP-10. Por lo tanto, el diseño basado en fragmentos que hemos utilizado ha resultado muy eficaz para obtener nuevos inhibidores selectivos de esta importante familia de enzimas.
4. CONCLUSIONES
Utilizando una aproximación de diseño basado en fragmentos, y el potencial de la química “click”, se ha llevado a cabo la síntesis de una serie de inhibidores potentes y selectivos de MMP-2. En primer lugar, se ha sintetizado un fragmento que contiene el grupo hidroxamato que actúa como Zinc Binding Group y un grupo azida necesario para la reacción CuAAC. Este fragmento se une a diferentes alquinos, seleccionados para unirse al bolsillo hidrófobo S1’ de la enzima. La actividad inhibitoria de los compuestos obtenidos frente a MMP-2 y MMP-9 se ha evaluado mediante un ensayo de zimografía. Se seleccionaron los dos compuestos con mejor perfil de inhibición para estudiar su actividad y selectividad frente a un panel de 10 metaloproteasas en un ensayo colorimétrico. Entre todos los inhibidores seleccionados cabe destacar el hidroxamato 20 con una actividad del orden nanomolar frente a MMP-2 y MMP-13 en este ensayo, mientras que es totalmente inactivo frente a MMP-1 y MMP-7 y 70 veces menos activo frente a MMP-9. Asimismo presenta una menor actividad frente a MMP-10 y MMP-14.
5. AGRADECIMIENTOS
Este trabajo ha sido financiado por el Ministerio de Ciencia e Innovación (SAF2005-02608, SAF2008-00945, y SAF2009-13240). Al Ministerio de Educación (programa FPU) por la beca a P. S., a la Fundación Universitaria San Pablo CEU por la beca de J. M. Z. y a la empresa EADS-CASA por la beca a K. F.
6. REFERENCIAS
1. Niebroj-Dobosz, I., & al. (2010). Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. European Journal of Neurology, 17(2), 226-231.
2. Yang, Y., & al. (2010). Increased intranuclear matrix metalloproteinase activity in neurons interferes with oxidative DNA repair in focal cerebral ischemia. Journal of Neurochemistry, 112(1), 134-149.
3. Zhou, H., & al. (2008). Sinomenine ameliorates arthritis via MMPs, TIMPs, and cytokines in rats. Biochemical and Biophysical Research Communications, 376(2), 352-357.
4. Nicolescu, A.C., & al. (2009). Inhibition of matrix metalloproteinase-2 by PARP inhibitors. Biochemical and Biophysical Research Communications, 387(4), 646-650.
5. Seo, K.W., & al. (2010). Participation of 5-lipoxygenase-derived LTB4 in 4-hydroxynonenal- enhanced MMP-2 production in vascular smooth muscle cells. Atherosclerosis, 208 (1).
6. Chetty, C., & al. (2008). Tissue inhibitor of metalloproteinase 3 suppresses tumor angiogenesis in matrix metalloproteinase 2-down-regulated lung cancer. Cancer Research, 68(12), 4736-4745.
7. Overall, C.M., & al. (2006). Towards third generation matrix metalloproteinase inhibitors for cancer therapy. British Journal of Cancer, 94(7), 941-946.
8. Binker, M.G., & al. (2009). EGF promotes invasion by PANC-1 cells through Racl/ROS- dependent secretion and activation of MMP-2. Biochemical and Biophysical Research Communications, 379(2), 445-450.
9. Overall, C.M., & O. Kleifeld (2006). Tumour microenvironment - Opinion - Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nature Reviews Cancer, 6(3), 227-239.
10. Xi, L., & al. (2009). A combined molecular modeling study on gelatinases and their potent inhibitors. Journal of Computational Chemistry, 31(1), 24-42.
11. Summers, J.B. (1998). in Annual Reports in Medicinal Chemistry, 33, D. Robertson and J. Plattner, Editors, Academic Press: San Diego, 131-149.
12. Terp, G.E., & al. (2000). Structural differences of matrix metalloproteinases. Homology modeling and energy minimization of enzyme-substrate complexes. Journal of Biomolecular Structure & Dynamics, 17(6), 933-946.
13. Bode, W., & al. (1999). Structural properties of matrix metalloproteinases. Cellular and Molecular Life Sciences: CMLS, 55(4), 639-652.
14. Natchus, M.G., & al. (2000). Development of new hydroxamate matrix metalloproteinase inhibitors derived from functionalized 4-aminoprolines. Journal of Medicinal Chemistry, 43(26), 4948-63.
15. Scozzafava, A., & al. (2000). Protease inhibitors: Synthesis of potent bacterial collagenase and matrix metalloproteinase inhibitors incorporating N-4- nitrobenzylsulfonylglycine hydroxamate moieties. Journal of Medicinal Chemistry, 43(9), 1858-1865.
16. Almstead, N.G., & al. (1999). Design, synthesis, and biological evaluation of potent thiazine- and thiazepine-based matrix metalloproteinase inhibitors. Journal of Medicinal Chemistry, 42(22), 4547-4562.
17. Pikul, S., & al. (1998). Discovery of potent, achiral matrix metalloproteinase inhibitors. Journal of Medicinal Chemistry, 41(19), 3568-71.
18. Kessenbrock., K, & al. (2010). Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell, 141(1), 52-67.
19. Tuccinardi, T., & al. (2006). Amber force field implementation, molecular modelling study, synthesis and MMP-1/MMP-2 inhibition profile of (R) and (S)-N-hydroxy-2-(N- isopropoxybiphenyl-4-ylsulfonamido)-3-methylbutanamid es. Bioorganic & Medicinal Chemistry, 14(12), 4260-4276.
20. Martin, M.D., & al. (2007). The other side of MMPs: Protective roles in tumor progression. Cancer and Metastasis Reviews, 26(3-4), 717-724.
21. Almholt, K., & al. (2008). Metastasis is strongly reduced by the matrix metalloproteinase inhibitor Galardin in the MMTV-PymT transgenic breast cancer model. Molecular Cancer Therapeutics, 7(9), 2758-2767.
22. Dashevsky., O, & al. (2009). Platelet-derived microparticles promote invasiveness of prostate cancer cells via upregulation of MMP-2 production. International Journal of Cancer, 124(8), 1773-1777.
23. Pfaffen, S., & al. (2010). Isolation and characterization of human monoclonal antibodies specific to MMP-1A, MMP-2 and MMP-3. Experimental Cell Research, 316(5), 836-847.
24. Whittaker., M, & al. (1999). Design and therapeutic application of matrix metalloproteinase inhibitors. Chemical Reviews, 99(9), 2735-2776.
25. Becker, D., & al. (2005). Synthesis and structure-activity relationships of beta- and alpha- piperidine sulfone hydroxamic acid matrix metalloproteinase inhibitors with oral antitumor efficacy. Journal of Medicinal Chemistry, 48(21), 6713-6730.
26. Kolodziej., S.A, & al. (2010). Orally bioavailable dual MMP-1/MMP-14 sparing, MMP-13 selective alpha-sulfone hydroxamates. Bioorganic & Medicinal Chemistry Letters, 20(12), 3557-3560.
27. Kolodziej, S.A., & al. (2010). MMP-13 selective isonipecotamide alpha-sulfone hydroxamates. Bioorganic & Medicinal Chemistry Letters, 20(12), 3561-3564.
28. Skiles, J.W., & al. (2004). The design, structure, and clinical update of small molecular weight matrix metalloproteinase inhibitors. Current Medicinal Chemistry, 11(22), 2911-2977.
29. Manetsch, R., & al. In situ click chemistry: Enzyme inhibitors made to their own specifications. Journal of the American Chemical Society, (2004). 126(40), 12809-12818.
30. Lewis, W.G, et al. (2002). Click chemistry in situ: Acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angewandte Chemie-International Edition, 41(6), 1053-1057.
31. Krasinski, A., & al. (2005). In situ selection of lead compounds by click chemistry: Target- guided optimization of acetylcholinesterase inhibitors. Journal of the American Chemical Society, 127(18), 6686-6692.
32. Whiting, M., & al. (2006). Inhibitors of HIV-1 protease by using in situ click chemistry. Angewandte Chemie-International Edition, 45(9), 1435-1439.
33. Giffin, M.J., & al. (2008). A Copper(I)-Catalyzed 1,2,3-Triazole Azide-Alkyne Click Compound is a Potent Inhibitor of a Multidrug-Resistant HIV-1 Protease Variant. Journal of Medicinal Chemistry, 51(20), 6263-6270.
34. Kalesh, K.A., & al. (2009). Rapid synthesis of Abelson tyrosine kinase inhibitors using click chemistry. Organic & Biomolecular Chemistry, 7(24), 5129-36.
35. Carvalho, I., & al. (2010). 'Click chemistry' synthesis of a library of 1,2,3-triazole- substituted galactose derivatives and their evaluation against Trypanosoma cruzi and its cell surface trans-sialidase. Bioorganic & Medicinal Chemistry, 18(7), 2412-2427.
36. Ng, S.L., & al. (2008). Click synthesis of small-molecule inhibitors targeting caspases. Organic & Biomolecular Chemistry.
37. Wang., J, & al. (2006). Rapid assembly of matrix metalloprotease inhibitors using click chemistry. Organic Letters, 8(17), 3821-3824.
38. GLIDE (2003). Glide v 2.5021. Schrödinger, L.L.C., New York, USA.
39. Feng, Y.Q., & al. (2002). Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. Biochimica Et Biophysica Acta-Proteins and Proteomics, 1598(1-2), 10-23.
40. Garcia, M.A., & al. (2007). Are adrenomedullin positive modulators novel matrix metalloproteinase inhibitors? Anales de la Real Academia de Farmacia; Instituto de España, 73, 703-724.
41. Garcia, M.A. (2007). Estudio de interacciones ligando-receptor mediante técnicas computacionales. La adrenomedulina y las metaloproteasas de la matriz como dianas para el diseño racional de fármacos. Tesis Doctoral Universidad San Pablo CEU.
42. Friesner, R.A., & al. (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem, 49(21), 6177-96.
43. MacPherson, L.J., & al. (1997). Discovery of CGS 27023A, a non-peptidic, potent, and orally active stromelysin inhibitor that blocks cartilage degradation in rabbits. J Med Chem, 40(16), 2525-2532.
44. Serra. (2009). Estrategias computacionales para el estudio de la inhibición de metaloproteasa de la matriz-2. Diseño de inhibidores. DEA Universidad San Pablo CEU.
45. Overall, C.M., & Lopez-Otin, C. (2002). Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nature Reviews Cancer, 2(9), 657-672.