REVISIÓN |
Lípidos y diabetes tipo 2. Desarrollo de nuevas terapias basadas en el metabolismo lipídico
José Carlos Rodríguez Rey
Departamento de Biología Molecular. Facultad de Medicina. Universidad de Cantabria. Avda. Cardenal Herrera Oria s/n 39011 Santander.
e-mail: rodriguj@unican.es
Recibido el 27 de agosto de 2013. An. Real Acad. Farm. Vol 79, Nº 3 (2013), pag. 412-433.
RESUMEN
La diabetes tipo 2 es una de las principales causas de morbilidad a nivel global. La enfermedad se desarrolla tras una fase más o menos prolongada en la que los tejidos presentan resistencia a insulina. En el músculo, la resistencia a insulina se manifiesta como un bloqueo a la entrada de glucosa, mientras que en el hígado da lugar a un aumento de la gluconeogénesis y de la síntesis de lípidos. En ambos tejidos la propia acumulación de lípidos es la principal causa de la resistencia, que se produce por un aumento de la cantidad de diacilgliceroles intracelulares. Estos activan diversas proteín quinasas de la familia PKC que a su vez interfieren con el proceso de señalización por insulina. En el hígado esta interferencia afecta parcialmente a la señalización. La insulina produce un aumento de la síntesis de lípidos, sin ser capaz de bloquear la gluconeogénesis. El resultado es una progresiva acumulación de lípidos que agrava el fenómeno de resistencia y que, de no revertirse, progresará hasta una diabetes tipo 2. La regulación a largo plazo de la síntesis de lípidos por insulina está mediada por el factor de transcripción SREBP-1c. La síntesis de éste está regulada por numerosos receptores nucleares, y la utilización de ligandos de estos últimos está proporcionando una nueva vía para la prevención y el tratamiento de la diabetes tipo 2. |
Palabras clave: Diabetes tipo 2; Quinasas; Hígado.
ABSTRACT
Lipid metabolism in type 2 diabetes and development of new lipid- based therapies
Type 2 diabetes is one of the major causes of global morbidity. The disease develops after a period of insulin resistance of variable length. In skeletal muscle, insulin resistance is characterized by an impairment of glucose transport whereas in liver there are increases in both gluconeogenesis and lipid synthesis. In both tissues lipid accumulation is responsible for insulin resistance because of increases in intracellular diacylglycerols. These activate several members of the PKC family of protein kinases, which, in turn, impair the insulin-signaling pathway. Insulin signaling is only partially affected in the resistant liver and thus insulin induces an increase in lipid synthesis without being able to block gluconeogenesis. As a consequence, lipids progressively accumulate and worsen insulin resistance, in a process, which, if not reversed, will result in an overt, type 2 diabetes. The transcription factor SREBP-1c is the major effector of long-term regulation of lipid synthesis by insulin. The synthesis of SREBP-1c is regulated by several nuclear receptors and the utilization of their ligands is opening a new therapeutic approach for the prevention and treatment of type 2 diabetes. |
Keywords: Type 2 diabetes; Kinases; Liver.
introducción
La diabetes tipo 2 es una de las principales causas de morbilidad en todo el mundo. Relativamente rara a principios de siglo XX, el aumento de su frecuencia está ligado al aumento de la obesidad y el sedentarismo. De cumplirse las proyecciones de la Organización Mundial de la Salud, en el año 2025 podría haber más de 350 millones de afectados por la enfermedad, por lo que no es exagerado considerarla como una de las epidemias del s. XXI (1). A diferencia de su homónima de tipo 1, de naturaleza autoinmune, la diabetes tipo 2 aparece tras un largo proceso. En el estado que puede denominarse pre-diabético, las células β del páncreas secretan un exceso de insulina para compensar una menor respuesta de los tejidos, y los niveles de glucosa se mantienen dentro de la normalidad. Cuando finalmente el páncreas no puede producir suficiente insulina para compensar la resistencia, los niveles de azúcar aumentan y la diabetes se manifiesta de forma franca (2).
El aumento de los niveles de azúcar en sangre es una de las características más llamativas de la diabetes y por esta razón la enfermedad ha sido asociada casi de forma exclusiva al metabolismo glucídico. El propio nombre de la enfermedad, diabetes mellitus, atribuido al médico griego Areteo (80-138) hace referencia al sabor dulce de la orina de los pacientes. Unos siglos antes (s. VI a.C.) los médicos indios ya la habían denominado Madhumeha, que significa literalmente orina-miel, y era frecuente la utilización de pruebas diagnósticas basadas en la atracción que las hormigas sentían por la orina dulce de los pacientes. Nombres similares pueden encontrarse en los ideogramas de los coreanos, chinos y japoneses. El sabor dulce de la orina se atribuye definitivamente a la presencia de cantidades elevadas de glucosa en 1776, cuando Matthew Dobson desarrolla un método para su cuantificación (3).
En opinión de algunos científicos la insistencia en el carácter glucídico de la enfermedad podría haber desviado la atención del importante papel de los lípidos en el desarrollo de la patología. En un artículo publicado en 1992 en la revista Science titulado “Si Minkowski hubiera sido agéusico. Una visión alternativa de la diabetes”, J. Denis McGarry especula con la posibilidad de que Minkowski, hubiera perdido la capacidad de distinguir el sabor dulce de la orina de los perros a los que previamente había convertido en diabéticos tras una pancreatectomía. En ausencia del sentido del gusto, razona McGarry, tal vez habría sido capaz de detectar el olor de los cuerpos cetónicos, algo que sin duda habría dirigido a la comunidad científica hacia el estudio del metabolismo de los lípidos (4).
El ciclo de Randle y el modelo lipogénico de la diabetes
A pesar del énfasis en el metabolismo de la glucosa, la relación entre los lípidos y la resistencia a insulina se conoce desde hace tiempo. Estudios llevados a cabo en músculos humanos han determinado que la presencia de lípidos en el interior de la célula muscular constituye el mejor predictor de la resistencia a insulina en ese tejido (5). De la misma forma, la resistencia a insulina hepática está muy relacionada con el contenido de lípido intrahepático, siendo este un mejor indicador que la propia adiposidad visceral (6)
Las primeras observaciones experimentales sobre las conexiones entre un aumento de los niveles de lípidos y la ralentización del metabolismo de la glucosa fueron publicadas por Randle, quien incubando preparaciones de músculo con ácidos grasos observó un incremento de los niveles de glucosa y glucosa 6-fosfato, así como un aumento de los depósitos de glucógeno intracelular. Basándose en estos resultados propuso un mecanismo de regulación alostérico consistente en la inhibición de la piruvato deshidrogenasa a causa del aumento de los cocientes acetil CoA/CoA y NADH/NAD+ en el interior de la mitocondria. El consiguiente incremento de citrato inhibiría dos pasos de la glucolisis: la conversión de glucosa a glucosa 6-fosfato, catalizada por las hexoquinasas y la formación de fructosa 1,6- bisfosfato, la reacción clave en la regulación de la glucolisis. Por su parte la glucosa 6-fosfato acumulada en ausencia de glucolisis sería derivada a la formación de glucógeno (Figura 1) (7, 8). Este mecanismo pudo ser demostrado en animales en los que infusiones de elevadas cantidades de lípidos en cortos períodos de tiempo producían un aumento de glucosa 6 –fosfato (9) y, así, la hipótesis de Randle se mantuvo como paradigma de la diabetes a lo largo de varias décadas.
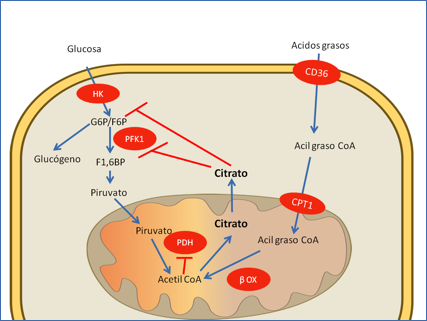
Figura 1.- Modelo alostérico de Randle para explicar la resistencia a insulina muscular tras la acumulación intracelular de ácidos grasos. Los ácidos grasos que entran a la célula a través de CD36 son transportados al interior de la célula por la lanzadera de carnitina. La producción de grandes cantidades de acetil CoA por la beta oxidación y la síntesis de citrato dan lugar a la inhibición de PFK1 y HK, y al bloqueo de la glucolisis. La G6P acumulada se deriva hacia la síntesis de glucógeno. Abreviaturas: CPT1 carnitina palmitil transferasa I; HK- hexoquinasa; PFK1, fosfofructoquinasa 1; PDH, piruvato deshidrogenasa;, transportador de ácidos grasos; β-ox, beta oxidación de ácidos grasos. Figura adaptada de la referencia (8).
Casi treinta años después de la propuesta de Randle, McGarry completa el modelo postulando que la desregulación de las rutas del metabolismo lipídico es probablemente el suceso inicial en el desarrollo de la diabetes (4). De acuerdo con McGarry, puesto que los niveles de lípidos del músculo son los causantes de la resistencia del mismo, la causa de la diabetes debería buscarse en las modificaciones del metabolismo que dan lugar a aumentos de los niveles de lípidos sanguíneos.
La síntesis y degradación de ácidos grasos se regula de forma recíproca gracias al papel de un metabolito clave, el malonilCoA, precursor de la síntesis de ácidos grasos. Producido por la acción de la acetil CoA carboxilasa, es a su vez un regulador de CPT-1, enzima clave del transporte de ácidos grasos al interior de la mitocondria para su degradación. Sobre la pareja Acetil CoA carboxilasa/CPT1 recaería por lo tanto todo el peso de la regulación (Figura 2). En el músculo, en el que la síntesis de ácidos grasos carece de importancia, el malonil CoA se produce por acción de un isoenzima específico asociado a la membrana externa de la mitocondria, en donde también se sitúa CPT-1, por lo que los cambios de su actividad serían rápidamente trasladables a la actividad del transportador. En el músculo resistente a insulina los niveles de malonil CoA están aumentados (10) lo que estaría de acuerdo con la importancia del isoenzima muscular, codificado por el gen ACACB y cuyas variantes se han asociado recientemente con la obesidad y la diabetes (11).
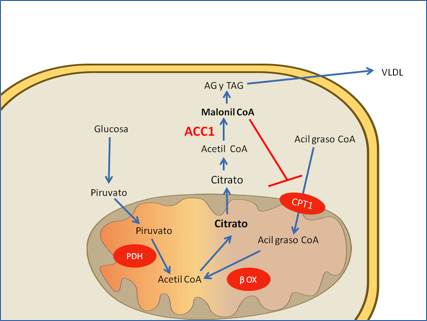
Figura 2.- Regulación recíproca de la síntesis y degradación de ácidos grasos a través de malonil CoA. El malonil CoA producido por acción de la acetil CoA carboxilasa inhibe la entrada de ácidos grasos a la mitocondria y como consecuencia su degradación. AG, ácidos grasos; Abreviaturas: ACC1 , acetil coA carboxilasa 1; CPT1 carnitina palmitil transferasa I; PDH, piruvato deshidrogenasaTAG, triacilgliceroles; VLDL lipoproteínas de muy baja densidad. β-ox, beta oxidación de ácidos grasos. Figura adaptada de la referencia (8).
La hipótesis lipogénica de la diabetes tipo 2 plantea una paradoja. Por una parte, las células musculares no utilizan glucosa por oxidar de preferencia ácidos grasos. Por otra, estas células acumulan ácidos grasos porque su capacidad de oxidarlos de forma eficiente está disminuida. Para resolverla, McGarry propone que el proceso de aparición de la diabetes se produciría en dos fases. En una fase temprana, previa al aumento de los niveles sanguíneos de ácidos grasos y triacilgliceroles, la célula muscular podría tener un defecto muy sutil - tal vez una desregulación del par Acetil CoA carboxilasa/CPT1- que le imposibilitara la degradación completa de los ácidos grasos. La acumulación de lípidos resultante daría lugar a la resistencia a insulina. Más adelante, cuando la secreción de insulina no pueda ya controlar la resistencia se produciría un aumento de los niveles de lípidos circulantes, cuya concentración sería ahora suficiente para vencer el bloqueo existente para su degradación. En esta fase la inhibición descrita por Randle entraría en juego para bloquear la degradación de glucosa muscular (12).
Considerando el importante papel del hígado y del tejido adiposo en el control de los niveles sanguíneos de lípidos, el proceso podría representarse en forma de un círculo vicioso (Figura 3). La liberación continuada de insulina resultante de una sobrealimentación daría lugar, entre otros efectos, a un aumento de la síntesis y liberación de lípidos por parte del hígado. Como consecuencia se produciría una acumulación de los mismos en hígado (esteatosis), músculo y tejido adiposo. El aumento de los lípidos intracelulares y por tanto de la resistencia a insulina, tanto en el hígado como en los tejidos periféricos, obligaría al páncreas a producir niveles crecientes de insulina de forma continuada. Una vez sobrepasada la capacidad del páncreas de hacer frente a estas demandas, los tejidos serían incapaces de normalizar los niveles de glucosa y se producirían niveles aumentados de glucosa incluso durante el ayuno.
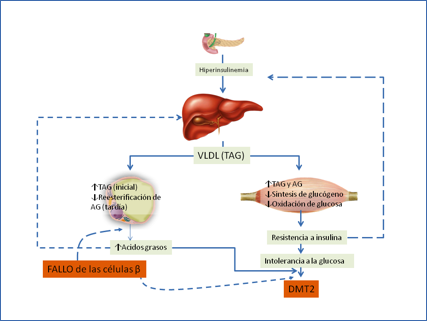
Figura 3.- Modelo lipogénico de la diabetes tipo 2 de McGarry. El aumento de la secreción de insulina producida por la sobrealimentación favorece la acumulación de lípidos en hígado, músculo y tejido adiposo, lo que su vez provocarán una mayor resistencia a insulina. Se produce así un círculo vicioso en el que páncreas compensa esta resistencia mediante la secreción de cantidades crecientes de insulina. Finalmente, el fallo de las células β provoca la rotura del círculo y la diabetes tipo 2 (4,12).
Alternativas a la regulación alostérica
Tanto el mecanismo propuesto por Randle como la modificación de McGarry se apoyan en el control alostérico que compuestos como el acetilCoA y malonil CoA ejercen sobre las rutas metabólicas. En el sistema de regulación metabólica de Randle la glucosa, que no se degrada en presencia de ácidos grasos, se acumularía en forma de glucosa 6- fosfato y glucógeno. Sin embargo, con el desarrollo de técnicas de análisis menos invasivas se pudo determinar que en el músculo de pacientes de diabetes hay una menor concentración de glucosa 6- fosfato y una síntesis de glucógeno disminuida a aproximadamente la mitad de la de los individuos sanos. El menor consumo de glucosa en los músculos de estos pacientes parece más el resultado de un menor transporte que de una menor degradación (13-15). Un efecto similar pudo también ser observado en individuos sanos sometidos a una infusión de lípidos y heparina. Esta, al inducir la lipoproteína lipasa, da lugar a una acumulación de lípidos en el interior de las células musculares (16). La resistencia a insulina observada en estos individuos se producía por los mismos mecanismos (disminución del transporte y de fosforilación de glucosa) observados en los pacientes de diabetes tipo 2 y sus familiares (17, 18). Todos estos sugerían que la regulación alostérica de la glucolisis propuesta por Randle, podría explicar los efectos que se producen tras un incremento agudo de los niveles de lípidos, pero no los cambios en el transporte de glucosa al interior de la célula.
El modelo por lo tanto debe ser modificado mediante la introducción de otro tipo de mecanismos que relacionen la presencia de lípidos con una menor entrada de glucosa en la célula muscular. A este respecto, no debería olvidarse que los estudios genómicos recientes han señalado que variantes en los genes que codifican proteínas relacionadas son los que presentan mejor asociación con la diabetes (19). Por lo tanto, un modelo completo de la diabetes que pretenda explicar los cambios en la homeostasis de la glucosa debería tomar en consideración (i) los mecanismos mediante los cuales la acumulación de lípidos afecta al mecanismo de señalización por insulina en células hepáticas, musculares y adipocitos, así como (ii) qué procesos dan lugar a una acumulación de lípidos en el interior de estas células.
Papel de la insulina en el transporte de la glucosa y en la síntesis de ácidos grasos
La insulina se secreta como respuesta a niveles elevados de glucosa en sangre, siendo el mantenimiento de los niveles sanguíneos de glucosa una de sus funciones. Además del páncreas, el hígado, el músculo y el tejido adiposo, son los principales tejidos involucrados en esta regulación. En presencia de insulina se activa la entrada de glucosa a tejido adiposo y músculo, la síntesis de glucógeno hepática y muscular, la síntesis hepática de ácidos grasos y triacilgliceroles y la acumulación de estos últimos en el tejido adiposo. Por el contrario, el descenso de los niveles de insulina que se produce como consecuencia de una disminución de la glucosa sanguínea da lugar a síntesis y liberación de glucosa por el hígado y la liberación de ácidos grasos por parte del tejido adiposo. Todos estos procesos están afectados en mayor o menor medida en el individuo diabético, pero la entrada de glucosa al músculo y la síntesis hepática de lípidos parecen tener una mayor importancia relativa.
La insulina participa a varios niveles en la regulación metabólica. La regulación de la disponibilidad de sustrato (transportadores), la velocidad de las reacciones enzimáticas alterando bien la modificación covalente de enzimas, o la síntesis de enzimas reguladores del metabolismo, son procesos regulados por insulina y en todos ellos la regulación se establece como consecuencia de la unión de la hormona a su receptor situado en la membrana externa de la célula. El receptor de insulina es un tetrámero compuesto por dos tipos de subunidades de las que uno tiene un dominio citoplasmático con actividad tirosina quinasa. La activación de esta se produce como consecuencia de la interacción de la insulina con su receptor, que transmite un cambio conformacional a estos dominios y ocasiona una autofosforilación de los residuos serina y tirosina de los mismos (20). En los cambios metabólicos relacionados con la resistencia a insulina están involucradas al menos seis proteínas citoplasmáticas que sufren una fosforilación en sus tirosinas, en respuesta a la interacción insulina-receptor. De todas ellas (IRS-1-4,Cbl y APS(21)), las cuatro proteínas de la familia IRS han recibido considerable atención desde el descubrimiento de su primer miembro (22). Los sustratos IRS1 e IRS2 desempeñan funciones esenciales en la retirada de glucosa por el músculo y el tejido adiposo. Aunque los ratones deficientes en cada uno de ellos presentan resistencia a insulina periférica, solamente los ratones IRS2-/- presentan fenotipo diabético (23, 24). Dado que IRS-2 parece desempeñar además un papel importante en la función de las células β, estos resultados apoyan la idea de que la combinación de defectos en los tejidos diana y en la producción de insulina es necesaria para el desarrollo de la enfermedad.
La fosforilación dependiente de insulina de residuos tirosina de las proteínas IRS genera sitios de anclaje para muchas proteínas que contienen dominios SH2, entre las que se encuentra una fosfatidil- inositol- 3- quinasa de tipo 1A, un enzima que ha sido implicado en numerosos procesos biológicos (25). Las fosfatidil- inositol- 3- quinasas de tipo 1A son heterodímeros formados por dos subunidades, p85 y p110 que constituyen las suunidades reguladoras y catalítica respectivamente. p85 tiene dos dominios SH2 que son capaces de unirse a la secuencias que contienen tirosinas fosforiladas en los IRS. La unión provoca así una estimulación alostérica de la subunidad p110 y la consiguiente síntesis de fosfatidil inositol 3,4,5 trifosfato. Este se acumula y atrae a la membrana moléculas con dominios de homología con plecstrina (PH), entre ellas la serina-treonina Akt (PKB), que es activada por otra proteína también atraída a la membrana: la proteína quinasa dependiente de 3-fosfoinosítidos, PDK1. Las diferentes isoformas de Akt son serina-treonina quinasas que ocupan uno de los principales nodos de los sistemas de señalización en células eucarióticas. Por ello interviene en multitud de procesos que incluyen la regulación metabólica, el crecimiento y la proliferación, la apoptosis, etc. (26).
Akt y entrada de glucosa al músculo
La entrada de glucosa al músculo esquelético se produce por la intervención de Glut4, un transportador independiente de ATP. Una vez en el interior de la célula la glucosa es fosforilada por la hexoquinasa y, en ausencia del enzima Glucosa-6-fosfatasa, la glucosa- 6- fosfato resultante es incapaz de abandonar la célula por lo que la asociación del transportador y la hexoquinasa pueden considerarse una verdadera trampa de glucosa. Hasta ahora y dada las diferentes afinidades por la glucosa del transportador y la hexoquinasa, la entrada de glucosa en la célula mediante el transportador se ha venido considerando el paso limitante en la retirada de glucosa por parte de ambos tejidos, aunque hay autores que cuestionan este modelo (27). Sea cual sea el modelo verdadero, Glut4 desempeña un papel importante en la entrada de glucosa a la célula muscular y adiposa y es una de las principales funciones afectadas por la resistencia a insulina.
La proporción de Glut4 presente en la membrana oscila entre un 1 y un 40% y depende de la cantidad de insulina presente (28, 29). La proteína Glut4 no presente en membranas se reparte entre el aparato de Golgi y un compartimento especializado denominado vesículas de almacenamiento de Glut4 (GSV en sus siglas inglesas) desde donde es transportada a la membrana tras la estimulación de la célula por insulina (30, 31). El transporte se produce mediante un proceso de exocitosis en el que participa un complejo ternario formado por las proteínas VAMP 2, SNAP23 y sintaxina 4 (32).
En el interior de las vesículas Glut4 forma complejos con al menos otras dos proteínas: una amino-peptidasa, regulada por insulina (IRAP), y AS160, que precisamente recibe su nombre por constituir un sustrato de Akt (Akt-substrate of 160 KDa). AS160 tiene 6 sitios de fosforilación por Akt (33) y la eliminación de estos por mutagénesis previenen la traslocación del transportador (34). Asimismo el tratamiento con un RNA de interferencia específico de AS160 produce un aumento de Glut4 en la membrana en el estado basal, aunque se sigue manteniendo la inducibilidad por insulina, por lo que no puede descartarse la existencia de algún otro mecanismo que actúe de forma sinérgica con la anterior (35). El papel de AS160 no se limita a la inducción por insulina, ya que AS160 es un sustrato de la proteína quinasa dependiente de AMP (AMPK) lo que podría explicar la mayor entrada de glucosa en el músculo que se produce durante el ejercicio.
Akt y el metabolismo hepático
El hígado, cuyo principal transportador, Glut2, es independiente de insulina, modifica los niveles de glucosa mediante su conversión en glucógeno o en ácidos grasos. Se sabe desde hace tiempo que la insulina regula la actividad de los principales enzimas reguladores de estos dos procesos, así como los de la gluconeogénesis mediante modificación covalente. Sin embargo, la principal regulación de los procesos que se desarrollan en un período de tiempo largo, como es el caso de la diabetes tipo 2, se lleva a cabo alterando la síntesis de los propios enzimas. En el metabolismo hepático la insulina participa en esta regulación a través de dos factores de transcripción: FoxO1 y SREBP-1c (Figura 4a).
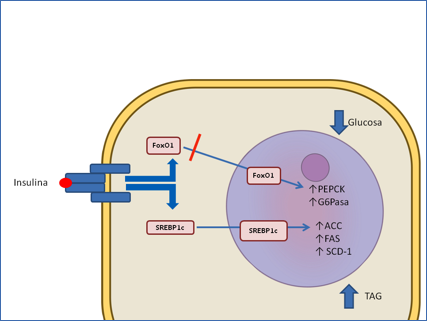
Figura 4a.- Regulación transcripcional de la gluconeogénesis y de la lipogénesis hepáticas por insulina. La interacción de la insulina con su receptor provoca la fosforilación del factor de transcripción FoxO1 y su salida del núcleo y retención en el citoplasma. Se inhibe así la expresión de los genes que codifican los principales enzimas reguladores de la gluconeogénesis, fosfoenol piruvato carboxi quinasa (PEPCK) y glucosa 6- fosfatasa (G6Pasa). La liberación e insulina da lugar también a un aumento de la síntesis y activación del factor de transcripción SREBP-1c, lo que resulta en una expresión aumentada de varios genes que codifican enzimas de la síntesis de ACC, acetil CoA carboxilasa; FAS, Sintasa de ácidos grasos; SCD-1, estearil CoA deshidrogenasa 1.
El factor de transcripción FoxO1 es un miembro de la familia de factores de transcripción Fox (Forkhead box) que interacciona con las secuencias denominadas elementos de respuesta a insulina (IRE) situadas en los promotores de numerosos genes. Entre sus dianas se encuentran los genes que codifican la fosfoenol- piruvato carboxiquinasa, el principal enzima regulador de la gluconeogénesis (36) y la glucosa 6 fosfatasa (G6P-asa). Mediante experimentos de ganancia y pérdida de función se ha podido demostrar que la presencia de FoxO1 aumenta la síntesis de glucosa en el hígado (38, 39). La insulina bloquea el proceso induciendo la fosforilación de FoxO1 en el núcleo, lo que no solamente excluye a FoxO1 de los complejos transcripcionales sino que además dispara los procesos de su envío al citoplasma (26, 40).
El otro factor de transcripción, SREBP-1c, pertenece a una familia de proteínas que comprende tres miembros con funciones muy relacionadas con el metabolismo de colesterol y ácidos grasos. El factor de transcripción inactivo es un dominio de una proteína más grande anclada en la membrana del retículo endoplásmico. SREBP-1c regula positivamente la transcripción de genes que codifican importantes proteínas de la síntesis de ácidos grasos y triacilgliceroles: la ya mencionada acetil CoA carboxilasa y la sintasa de ácidos grasos, así como la estearil CoA deshidrogenasa (41, 42). La regulación de SREBP-1c es compleja, puesto que involucra tanto la regulación transcripcional como el procesamiento proteolítico de la proteína anclada a la membrana del retículo, siendo ambos procesos inducidos por insulina (42).
La comparación de los ratones deficientes en el receptor de insulina hepático (LIRKO) con pacientes de diabetes o con los modelos murinos de la enfermedad demostró que, aunque la gluconeogénesis y de la síntesis de lípidos son regulados por insulina, la regulación de ambos es independiente a partir de un cierto punto. En los ratones LIRKO, como consecuencia de la ausencia del receptor, no se produce ni la fosforilación de FoxO1 ni la activación de SREBP-1c. Como resultado la insulina no es capaz ni de inhibir la gluconeogénesis ni de activar la síntesis de triacilgliceroles. Así, los ratones presentan hiperglucemia e hiperinsulinemia, pero no triacilgliceroles aumentados (43, 44). Por el contrario en los pacientes de diabetes, y en los modelos murinos de la misma, la resistencia a insulina se manifiesta de forma parcial, inactivándose el bloqueo de FoxO1 y de la gluconeogénesis sin modificar la inducción de la síntesis de lípidos por SREBP-1c (Figura 4b) (45). Por tanto, los diabéticos tipo 2 presentan hiperlipidemia, además de la hiperglucemia e hiperinsulinemia característica de los ratones LIRKO. Paradójicamente, este fenómeno de resistencia parcial a la insulina da lugar a un fenotipo mucho más grave que la resistencia total desarrollada por los ratones LIRKO (46).
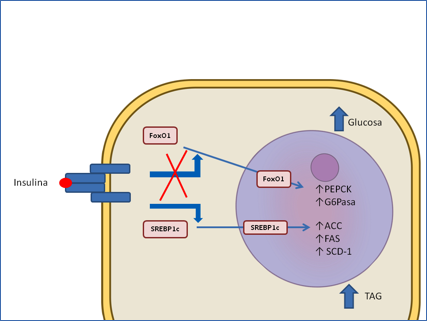
Figura 4b.- En el hígado diabético se produce un bloqueo en el control de la gluconeogénesis por insulina que no afecta a la inducción de la síntesis de ácidos grasos y triacilgliceroles. Adaptado de (46).
El punto en donde se produce la bifurcación de la regulación de FoxO1 y SREBP-1c ha sido identificado recientemente. Mientras que FoxO1 es un sustrato directo de Akt (revisado en (47)), la inducción de SREBP-1c por insulina, aunque también dependiente de Akt, requiere asimismo la participación del complejo mTORC1. De hecho, la rapamicina, inhibidor específico del complejo mTORC1- y de quien este recibe su nombre- es capaz de bloquear selectivamente la inducción de SREBP-1c por insulina (48). Este mecanismo proporciona además nexos de unión con otras situaciones metabólicas, puesto que se ha determinado de mTORC1 podría estar ligado a la obesidad a través de un mecanismo de estabilización en el que participa Notch (49). De la misma forma que la entrada de glucosa en el músculo, la actividad de SREBP-1c está sometida a regulación por la proteína quinasa dependiente de AMP, lo que de nuevo podría establecer una relación entre el ejercicio y la disminución de la resistencia hepática a insulina (50).
Mecanismos de resistencia a insulina mediada por lípidos
La identificación del tipo de lípidos responsables de la resistencia a insulina en el músculo ha sido un objeto de investigación muy activo en los últimos años. El hecho de que constituyan los principales componentes de los depósitos intracelulares de lípidos, podría señalar a los triacilglieroles como los principales responsables. Sin embargo, los ratones que sobreexpresan diacil-glicerol acil transferasa 1, el enzima que convierte diacilgliceroles en triacilgliceroles, presentan niveles de triacilgliceroles en músculo mucho más elevados sin alterar la sensibilidad a la insulina (51). Por su parte, en humanos es conocida la existencia de grandes depósitos de triacilgliceroles en los músculos de atletas entrenados, que son sujetos caracterizados por poseer una gran sensibilidad a insulina (52).
La utilización de animales modificados genéticamente ha permitido reconocer el papel de muchos de los enzimas del transporte, síntesis y degradación de lípidos en la aparición de la resistencia (para una discusión más detallada ver (53)), lo que ha llevado a identificar a los diacilgliceroles y las ceramidas como principales lípidos responsables de la resistencia, aunque el papel de éstas últimas podría depender de la presencia de grasas saturadas (54).
Los mecanismos de inhibición de la señalización por diacilgliceroles parecen estar mediados por quinasas de la familia PKC. Estudios iniciales demostraron que el acetato de forbol, un conocido activador de PKC, es capaz de producir resistencia a insulina (55). La participación de PKC está apoyada por la existencia de una activación crónica de PKC en modelos murinos de diabetes (56). Sin embargo, la resistencia a insulina no parece ser la consecuencia de la activación de un solo tipo de PKC. En el músculo, la infusión de lípidos con heparina, provoca un aumento de diacilgliceroles intramusculares y un bloqueo de la señalización por insulina asociada a la activación de PKCθ (57). Esto estaría de acuerdo con el hecho de que ratones deficientes en PKCθ están protegidos de la resistencia a insulina muscular inducida por este mismo mecanismo (58). Sin embargo, tras 14 semanas de alimentación rica en grasas, estos ratones presentan acumulación de lípidos en hígado, músculo y tejido adiposo, acompañados de la resistencia correspondiente (59). A la explicación de este fenómeno podría contribuir la existencia de al menos otros dos miembros de la familia PKC involucrados en las interferencias producidas por diacilgliceroles en el hígado. La resistencia a insulina hepática está asociada de forma específica a la activación de PKCε y los ratones deficientes no desarrollan resistencia a insulina como consecuencia de dieta rica en grasa, a pesar de desarrollar esteatosis hepática (60). A este efecto podría sumarse la resistencia hepática mediada por PKCδ. La activación de esta por una infusión de lípidos e insulina va acompañada de la activación de IKK-β, algo que podría conectar la resistencia a insulina con el fondo proinflamatorio que acompaña a la hiperlipidemia crónica (61). Asimismo, el estado proinflamatorio que acompaña a la obesidad podría estar conectado a la resistencia a través de la inducción de PKCζ por ceramidas, de acuerdo a un mecanismo en que podrían participar los ácidos grasos saturados como posibles ligandos del receptor TLR4 (62). Por otra parte, las ceramidas podrían activar la proteína fosfatasa PP2A, que interrumpiría la vía desfosforilando Akt (63) (Figura 5).

Figura 5.- Esquema de las principales vías de regulación que participan en la resistencia a insulina y principales puntos de inhibición por diacilgliceroles, ceramidas y citoquinas pro-inflamatorias por lípidos. Para una explicación más detallada del mecanismo consultar el texto.
El mecanismo concreto mediante el cual las quinasas de la familia PKC bloquean la señalización por insulina no está totalmente establecido. Existe un consenso en que la fosforilación de componentes de la ruta podría ser la causante del bloqueo, aunque los puntos concretos en que se produce están por determinar. El receptor de insulina, una posible diana, posee varios posibles sitios de fosforilación por PKC, lo que sugiere un mecanismo de bloqueo de la señal. En el hígado de rata, PKCε y el receptor de insulina se encuentran muy próximos y la resistencia podría ser el resultado de su interacción directa. No obstante, el análisis de biopsias de músculo de pacientes diabéticos no ha podido demostrar esta fosforilación in vivo(64). Se ha propuesto también que la fosforilación de la Ser 1101 de IRS-1 por parte de PKCθ, podría impedir su fosforilación por el propio receptor (65). En el caso de la inducción de PKCζ por ceramidas, se sabe que PKCζ y la isoforma de Akt más abundante en hígado, Akt2, son capaces de interaccionar. Siguiendo un mecanismo parecido a los mencionados para el receptor e IRS-1, la fosforilacion de Akt por PKCζ podría impedir su activación por PDK1 (66) (Figura 5).
La regulación transcripcional de SREBP- 1c como diana terapeútica
De acuerdo con el modelo mostrado en la Figura 3, la diabetes es la consecuencia del círculo vicioso que forman la sobreproducción de lípidos en el hígado y la sobreproducción de insulina por el páncreas. Siguiendo este razonamiento, la reducción de la síntesis hepática de ácidos grasos podría ayudar a revertir el proceso y aumentar la sensibilidad a insulina. Si además se considera la importancia en la síntesis de grasos de la regulación transcripcional por SREBP-1c, tanto la disminución de la síntesis o activación de este podría resultar efectiva en el tratamiento de la diabetes.
Se conocen diversos ejemplos de compuestos que son capaces de inhibir la expresión de SREBP-1c, entre ellos algunos productos naturales presentes en la uva o en el aceite de pescado (72, 73). Algún fármaco antidiabético, como la metformina, posee también este efecto, al igual que AICAR, un inhibidor de la proteína quinasa dependiente de AMP , aunque al poseer ambos este último efecto, es difícil atribuir el efecto antidiabético exclusivamente a la regulación de SREBP-1c (50).
Los receptores nucleares pueden desempeñar un papel importante en la regulación de los procesos que conducen a la resistencia a insulina y, de hecho, algunos de sus ligandos se utilizan para el tratamiento de la diabetes. Desde hace bastante tiempo se conoce el efecto diabetógeno de los glucocorticoides, cuyo receptor disminuye la secreción de insulina por el páncreas, así como la síntesis de componentes esenciales de la cascada de señalización (67). Asimismo, los receptores de oxisteroles (LXR) favorecen la formación de hígado graso no alcohólico, probablemente vía un aumento de la expresión de SREBP-1c (68). Paradójicamente, algunos agonistas sintéticos de LXR pueden tener efecto antidiabético, un efecto que se ha atribuido a un aumento de la expresión de Glut4 en los tejidos periféricos(69). Por el contrario, los fibratos y las tiazolidinodionas, agonistas de PPAR alfa y gamma respectivamente, tienen un efecto beneficioso sobre la sensibilidad a insulina y son ampliamente utilizados en clínica (70, 71). Aunque en muchos casos solamente se conoce el efecto de los receptores nucleares en los modelos murinos, su acción se mantiene en aquellos que ya han sido trasladados a modelos humanos (Tabla 1).
Tabla 1.- Efectos conocidos de receptores nucleares sobre la resistencia a insulina. +, evidencia de efectos antidiabéticos; - evidencia de efectos prodiabéticos. ?. No hay evidencia o la existente no es concluyente. Adaptado de (69).
Receptor nuclear |
Modelos murinos |
Pruebas en pacientes |
GR |
- |
- |
LXRα/β |
+/- |
? |
FXR |
+/- |
+/? |
PPARα/γ/δ |
+/+/+ |
+/+/? |
ERβ |
+ |
+ |
CAR |
+ |
+ |
HNF4-α |
+ |
+ |
VDR |
+ |
? |
TRβ |
+ |
? |
NR4A1-3 |
+ |
? |
NR5A2 (LRH-1) |
+ |
? |
Aunque el efecto de los receptores nucleares sobre la expresión de SREBP-1c es conocido desde hace tiempo, la utilización terapeútica de ligandos de receptores nucleares con el objetivo específico de disminuirla es relativamente reciente. La disminución de SREBP-1c podría explicar la sensibilización a insulina obtenida tras tratamiento con agonistas de CAR(74) y NR5A2(75). La activación de ambos receptores nucleares reprime la expresión de SREBP-1c, y por lo tanto de sus dianas lipogénicas. Al menos en el caso de CAR, la reducción en la síntesis de ácidos grasos vendría acompañada de un aumento de la degradación de los mismos presumiblemente a través de la disminución de la síntesis de malonil CoA. Algunos de estos compuestos, como el DLPC, un fosfolípido natural ligando de NR5A2, que están siendo ensayados para su aplicación en clínica (76), podrían abrir una nueva vía (Figura 6) para el tratamiento de la resistencia a insulina y la diabetes tipo 2.
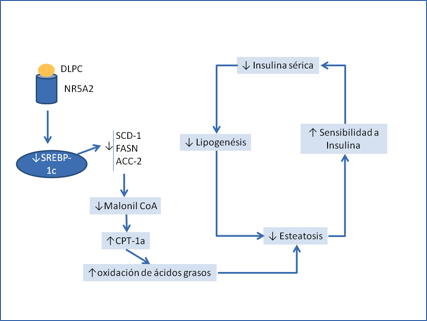
Figura 6.- Ejemplo de posible aplicación de los ligandos de receptores nucleares al tratamiento de la resistencia a insulina y la diabetes tipo 2. El fosfolípido DLPC (dilauril-fosfatidil colina) funciona como ligando del receptor nuclear NR5A2. Al unirse a su receptor regulará negativamente la expresión del gen que codifica SREBP-1c, lo que producirá a su vez una disminución de la expresión de los genes que codifican los enzimas de la síntesis de lípidos. Al disminuir la expresión de acetil CoA carboxilasa, disminuirá la síntesis de malonil coA y eso permitirá una mayor entrada de ácidos grasos a la mitocondria para su degradación. La conjunción de una menor síntesis y una mayor degradación hará que disminuyan los lípidos intracelulares y como consecuencia la resistencia a insulina. Se revertiría así el círculo vicioso descrito en la figura 3. Adaptado de (69).
REFERENCIAS
1. Hossain, P., Kawar, B., and El Nahas, M. 2007. Obesity and diabetes in the developing world--a growing challenge. N Engl J Med 356:213-215.
3. Polonsky, K.S. 2012. The past 200 years in diabetes. N Engl J Med 367:1332-1340.
19. McCarthy, M.I. 2010. Genomics, type 2 diabetes, and obesity. N Engl J Med 363:2339-2350.
25. Cantley, L.C. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655-1657.
71. Lehrke, M., and Lazar, M.A. 2005. The many faces of PPARgamma. Cell 123:993-999.
72. Del Bas, J.M., Ricketts, M.L., Baiges, I., Quesada, H., Ardevol, A., Salvado, M.J., Pujadas, G., Blay, M., Arola, L., Blade, C., et al. 2008. Dietary procyanidins lower triglyceride levels signaling through the nuclear receptor small heterodimer partner. Mol Nutr Food Res 52:1172-1181.