REVISIÓN |
Nuevas estrategias en terapia antitumoral basadas en la inducción de la apoptosis
Cristina Martín Sabroso1 y Ana Isabel Torres Suárez1,2*
1Departamento de Farmacia y Tecnología Farmacéutica, Facultad de Farmacia, Universidad Complutense de Madrid, Plaza Ramón y Cajal s/n, 28040, Madrid. 2Instituto de Farmacia Industrial. Universidad Complutense de Madrid.
e-mail: galaaaa@farm.ucm.es
Recibido el 3 de abril de 2013 An. Real Acad. Farm. Vol 79, Nº 2 (2013), pag. 213-228
RESUMEN
Según la OMS para el año 2030 se producirán un total de 11,5 millones de defunciones por cáncer. Una alta proporción de estos tumores serán resistentes a los agentes antineoplásicos actuales, por ello, en los últimos años se ha realizado una intensa labor investigadora en la búsqueda de nuevas dianas para el tratamiento de tumores. Una de las dianas que está ofreciendo más posibilidades es el mecanismo de la apoptosis. La apoptosis se produce en condiciones fisiológicas como un mecanismo regulador del crecimiento de tejidos, en equilibrio con la proliferación celular. Su desregulación podría ayudar a explicar la patogénesis de algunos tumores malignos. |
Palabras clave: Apoptosis; Antineoplásicos; Resistencia a fármacos.
ABSTRACT
New strategies in antitumor therapies based on apoptosis induction
According to OMS, in 2030 will be a total of 11.5 million deaths by cancer. A high proportion of these tumors will be resistant to current anticancer agents, and therefore, in the last years an intense investigation has been done to search new targets for antitumor treatment. One of the most interesting targets is the apoptosis mechanism. Apoptosis is produced in physiological conditions as a growth tissue regulator mechanism, in equilibrium with cellular proliferation. Its deregulation could explain the pathogenesis of some malignant tumors. |
Keywords: Apoptosis; Anticancer agent; Drug resistant.
1. Introducción
Según la OMS para el año 2030 el número de nuevos pacientes diagnosticados de cáncer ascenderá a 15,5 millones y se producirán un total de 11,5 millones de defunciones. Una alta proporción de estos tumores serán resistentes a los agentes antineoplásicos actuales, y por ello hoy en día hay numerosas líneas de investigación abiertas para desarrollar nuevos fármacos que superen estas resistencias.
La resistencia a los antineoplásicos puede ser natural o adquirida. La resistencia natural hace referencia a la baja respuesta inicial de un tumor a un determinado fármaco, mientras que la adquirida se refiere a la falta de respuesta que surge tras un tratamiento inicial con éxito. Existen tres características tumorales que parecen determinar la resistencia a la quimioterapia: la cinética de crecimiento, la aparición de mutaciones espontáneas y la bioquímica.
1. Cinética de crecimiento de la masa tumoral: La mayoría de los quimioterápicos actúan sobre las células en división, de manera que cuanto menor sea la fracción en crecimiento, es decir, el porcentaje de células con actividad proliferativa, menor será el número de células presumiblemente sensibles al tratamiento. Por ello, las células que se encuentran fuera del ciclo celular, en la llamada fase G0, serán refractarias a la mayoría de quimioterápicos (1). En los tumores sólidos, el crecimiento se enlentece de forma exponencial, aumentando el número de células en fase G0 según progresa el tumor. Una importante consecuencia de este tipo de crecimiento es que, en el momento del diagnóstico, la mayoría de los pacientes presentan tumores con una baja fracción de crecimiento y, por tanto, con baja sensibilidad a la mayoría de los quimioterápicos.
2. Aparición de mutaciones espontáneas: A través de cambios citogenéticos aleatorios, algunas células tumorales mutan volviéndose resistentes a ciertos fármacos, sin haber tenido contacto previo con ellos. Esta capacidad de resistencia se transmite a las células descendientes, de tal forma que la población celular de un tumor en crecimiento no es homogénea, coexistiendo clones celulares sensibles y resistentes al tratamiento. El número de células resistentes en un tumor en el momento del diagnóstico, se relaciona directamente con su tamaño y su tasa intrínseca de mutación (2,3).
3. Alteración de mecanismos bioquímicos: Pueden aparecer resistencias a quimioterápicos por diferentes razones bioquímicas que incluyen la incapacidad de que en la célula tumoral se transforme el profármaco a su forma activa, la capacidad de la célula tumoral de inactivar el fármaco(modificación del metabolismo intracelular del fármaco), la disminución de la captación del fármaco por parte de la célula tumoral (modificación de barreras fisiológicas, vascularización tumoral alterada…), el aumento de la expulsión del fármaco a través de la membrana celular (bombas de eflujo), cambios en los niveles o en la estructura de la diana intracelular, o el aumento de la tasa de reparación del ADN dañado.
Actualmente, se están utilizando diferentes estrategias para aumentar la respuesta a los antineoplásicos. Entre ellas se encuentra la detección precoz del tumor ya que si el tumor es de menor tamaño tendrá un mayor porcentaje de células en división y por tanto sensibles al tratamiento. En este sentido, también resulta eficaz la reducción de la masa tumoral mediante cirugía y/o radioterapia, ya que esta reducción aumenta la fracción de células en crecimiento y produce una mayor susceptibilidad a los fármacos.
Es especialmente importante la eliminación de las masas tumorales necróticas a las que, por baja vascularización, no puede llegar suficiente cantidad de fármaco. Para evitar las resistencias debidas a alteraciones bioquímicas se intenta favorecer el acceso del fármaco al tumor desarrollándose sistemas de vectorización. Mediante esta estrategia se persigue una localización selectiva del fármaco a nivel de la célula tumoral reduciéndose, de forma paralela, la toxicidad asociada a estos fármacos.
Se han desarrollados novedosos sistemas que permiten la vectorización pasiva o activa del fármaco a nivel de las masas o incluso el interior de las células tumorales, evitando en muchos casos, los mecanismos naturales de internalización de ese fármaco en la célula. La asociación de quimioterápicos es un recurso también muy utilizado, que ofrece dos ventajas: la suma o potenciación de efectos citotóxicos (se asocian moléculas con diferente mecanismo de acción), y la disminución de la probabilidad de aparición de resistencias (se asocian fármacos con diferentes mecanismos de resistencias).
Por último, en los últimos años se ha realizado una intensa labor investigadora en la búsqueda de nuevas dianas para el tratamiento de tumores que, por un lado resulten eficaces para la eliminación o detención del crecimiento tumoral y que por otro eviten o dificulten la aparición de resistencias. Una de las dianas que está ofreciendo más posibilidades es el mecanismo de la apoptosis. A continuación se presenta una revisión de las más recientes investigaciones en tratamiento antitumoral basadas en el desarrollo de moléculas que actúan sobre el mecanismo de la apoptosis, analizándose la posibilidad de aparición de resistencias y el balance de su aplicación clínica.
2. Mecanismo de apoptosis
La apoptosis, o muerte celular programada, es un modo específico de muerte, que se caracteriza por cambios morfológicos tales como condensación de la cromatina, la fragmentación del núcleo, la retracción citoplasmática y la emisión de 'cuerpos apoptóticos', manteniendo aparentemente intactos los orgánulos celulares (4,5). De hecho, la apoptosis regula un mecanismo de muerte celular que implica la participación activa de la célula afectada en la ejecución de su propio programa de muerte (Figura 1). Los eventos morfológicos y bioquímicos de la apoptosis se conocen desde hace mucho tiempo, y, en algunos aspectos, difieren de aquellos que llevan a la necrosis (6). La apoptosis se produce en condiciones fisiológicas como un mecanismo regulador del crecimiento de tejidos, en equilibrio con la proliferación celular (7). La apoptosis se ha convertido en un foco de interés en oncología debido a que su desregulación podría ayudar a explicar la patogénesis de algunos tumores malignos (8).
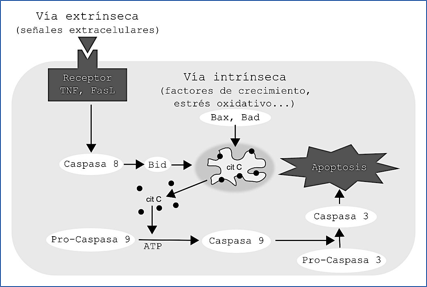
Figura 1.- Mecanismo general de activación de la apoptosis.
3. Inductores de la apoptosis
Se han identificado diferentes moléculas capaces de modificar el proceso de la apoptosis en las células tumorales, y que presentan interés como antineoplásicos (Tabla 1). Estos fármacos actúan sobre diferentes dianas moleculares y como consecuencia bien inducen una activación directa de la apoptosis, o bien reducen el umbral de su iniciación por fármacos citotóxicos (9).
Tabla 1.- Fármacos inductores de apoptosis con interés en el tratamiento del cáncer.
Inhibidores de la glutatión S-tranferasa |
Telcyta |
6-(7-nitro-2,1,3-benzoxadiazol-4-iltio)hexanol |
|
Inhibidores de mtor |
Temsirolimus |
Everolimus |
|
Deforolimus |
|
Inhibidores de EGFR |
Genitinib |
Erlotinib |
|
Cetuximab |
|
Matuzumab |
|
Inhibidores de la tubulina |
DJ-927 |
ABT-751 |
|
EPO-906 |
|
TZT-1027 |
|
Otros |
Fenoxodiol |
Inhibidores de c-kit y PDGFR |
3.1. Inhibidores de la glutatión S-transferasa (GST)
La glutatión S-transferasa(GST) representa el mayor grupo de enzimas de detoxificación, participando en la fase II de la biotransformación de compuestos tóxicos, exógenos o endógenos, mediante la catalización de reacciones de conjugación en las que se adiciona un grupo polar, el glutatión, a los grupos electrofílicos del tóxico a eliminar, entre los que se incluyen sustancias carcinógenas, dando como resultado un incremento de su solubilidad en agua que favorece su excreción renal.
La GST está también implicada en la regulación de la respuesta de estrés celular y apoptosis, debido a que provoca la inhibición de quinasas, como por ejemplo la jun N-terminal quinasa (JNK). Se han detectado niveles elevados de GST en cáncer de colon, vejiga, estomago, piel, mama, pulmón, bucal y riñón (10,11), en donde forma un complejo con JNK que provoca su inhibición. El estrés oxidativo puede desestabilizar el complejo GST:JNK y causar la activación de la cascada de quinasas que desencadena la apoptosis (12). Esta enzima, también se encuentra implicada en el desarrollo de resistencias de tumores frente diversos fármacos anticancerígenos. Por tanto el uso de inhibidores específicos de la enzima podría potenciar la eficacia de la quimioterapia sobre tumores resistentes a drogas anticancerígenas.
Esta enzima es la diana de algunos de los nuevos agentes antitumorales desarrollados como Telcyta y 6-(7-nitro-2,1,3-benzoxadiazol-4-iltio) hexanol (NBDHEX). El primero es un profármaco sustrato de la enzima, aprovechando su alta concentración a nivel tumoral; sin embargo el segundo es un inhibidor de la GST.
Canfosfamide HCl (Telcyta)
Canfosfamide HCl (Telcyta®) fue diseñado para aprovechar los altos niveles de GST en muchos tumores humanos, lo cual, frecuentemente se asocia con un mal pronóstico y resistencia a ciertos fármacos como agentes alquilantes y compuestos de platino. Canfosfamide HCl (CAN) es un profármaco análogo del glutation que es activado por GST e induce apoptosis. Tras la activación, la actividad apoptótica de canfosfamide está mediada por la vía de respuesta al estrés, lo que resulta en la inducción de la apoptosis celular. Las células tumorales humanas expuestas a canfosfamide muestran una activación de proteínas quinasas activadas por mitógenos (MAP), quinasa MKK4, la quinasa p38, jun N-terminal quinasa (JNK) y la caspasa 3 (13). La actividad citotóxica de canfosfamide se ha demostrado in vitro e in vivo frente una gran variedad de líneas celulares de cáncer humano. Canfosfamide generalmente, es bien tolerado y presenta pocos efectos adversos.
Canfosfamide ha demostrado no tener resistencia cruzada con platinos, taxanos y antraciclinas, tampoco su toxicidad se solapa con la de estos agentes pero sí posee un efecto sinérgico que hace que estos funcionen mejor en combinación que como agente único. Canfosfamide como agente único no es lo suficientemente activo por ello debe emplearse siempre en combinación con platinos, taxanos y antraciclinas.
6-(7-nitro-2,1,3-benzoxadiazol-4-iltio) hexanol (NBDHEX)
Un fármaco inhibidor de la GST es 6-(7-nitro-2,1,3-benzoxadiazol-4-iltio) hexanol (NBDHEX). Es un agente hidrófobo que penetra fácilmente a través de la membrana celular y por lo tanto, no es sustrato de las proteínas transportadoras (bombas de eflujo), lo que representa un mecanismo de resistencia del tumor a una variedad de fármacos anticancerosos. NBDHEX posee un importante efecto inhibitorio del crecimiento del melanoma maligno y de otros tipos de tumores, incluidos la leucemia, el cáncer de pulmón y el osteosarcoma (14,15). Este compuesto interrumpe el complejo entre GST y c-Jun N-terminal quinasa (JNK) induciendo la activación de JNK, la detención del ciclo celular y muerte celular (16). Ensayos clínicos indican que la combinación NBDHEX y temozolomida (TMZ) (agente alquilante) redujo significativamente el crecimiento tumoral in vivo con respecto a cada medicamento usado de forma individual, sin empeoramiento de la mielotoxicidad del agente alquilante ni retrasó la recuperación de la función de la médula ósea (17).
Las células de melanoma B16 fueron expuestas a diferentes concentraciones de NBDHEX en monoterapia, y los resultados indicaron que este agente tenía efecto apoptótico en las células B16 y que a una concentración de 10 µM inducía apoptosis en casi el 100% de las células a las 48 h.
3.2. Inhibidores de mtor
mTOR1 (mammalian target of rapamycin), es una serina/Treonina quinasa de 289 kDa, que participa en la vía fosfatidilinositol 3'-quinasa/AKT. Regula múltiples eventos celulares como el crecimiento celular y la proliferación, la diferenciación, la migración, y la supervivencia. Esta proteína está activada en múltiples tumores y su activación es capaz de generar la proliferación celular y de evitar la apoptosis, lo que conduce a su crecimiento desordenado. La rapamicina es un inhibidor de mTOR que actúa formando inicialmente un complejo con una proteína citosólica de 12 kDa, designado FK-506-proteína de unión 12 (FKBP-12), y posteriormente este complejo se une a mTOR inhibiendo su función, lo que desencadena una activación rápida y sostenida de la apoptosis mediante la vía de señalización de la quinasa 1 (18). De esta forma la rapamicina y sus análogos, temsirolimus, RAD001 y AP23573, ahora en ensayos clínicos como agentes anticancerosos, inhiben potentemente la proliferación de células tumorales.
Temsirolimus
Temsirolimus (CCI-779), es un éster derivado de rapamicina con mayor solubilidad en agua. A dosis no tóxicas, temsirolimus presentó actividad antitumoral sobre diversos modelos de cáncer tales como gliomas, rabdomiosarcomas, meduloblastoma, tumores de cabeza y cuello, líneas celulares de páncreas, próstata y mama (19), obteniéndose un mayor efecto terapéutico cuando se utilizó en combinación con los agentes antineoplásicos que por separado. En 2007 fue aprobado por la FDA como primera línea de tratamiento para el cáncer renal. También se han realizado estudios en fase III para el tratamiento de linfoma y en fase II en cáncer de mama y linfoma mostrando un beneficio frente a otros tratamientos especialmente en el caso del linfoma (20). Los principales efectos secundarios detectados son alteraciones hematológicas, hipercolesterolemia y reacciones alérgicas.
Everolimus
Everolimus (RAD001) es un derivado de la rapamicina de administración oral. Se ha evaluado su actividad antineoplásica en varias líneas celulares tumorales humanas in vitro y en modelos de xenoinjertos in vivo con una IC50 que va de 5 hasta 1800 nM. Se ha demostrado su efecto antineoplásico frente a melanoma, cáncer de pulmón, adenocarcinoma pancreático y carcinoma de colon. Este fármaco posee también actividad antiangiogénica ya que inhibe la proliferación de las células endoteliales vasculares humanas. Los efectos secundarios detectados a dosis terapéuticas son fatiga, neutropenia, mucositis, hiperglucemia y estomatitis (21). En un estudio en fase II, everolimus ha demostrado actividad antineoplásica en pacientes recién diagnosticados de cáncer de mama. También está siendo evaluado en ensayos con pacientes con cáncer de próstata y neuroendocrinos.
Deforolimus
Deforolimus (AP23573) es el último análogo desarrollado a partir de la rapamicina. Es estable en solventes orgánicos, soluble en agua en un amplio intervalo de pH, así como también en plasma y en sangre, tanto in vivo como in vitro. AP23573 presenta una potente, rápida y prolongada actividad inhibitoria de mTOR, lo que produce una inhibición del crecimiento de diversas líneas tumorales humanas in vitro, ejerciendo este efecto tanto individualmente como en combinación con otros agentes citotóxicos. En un estudio clínico de fase I, se administró por vía intravenosa durante 5 días en intervalos de 24 horas. Durante el primer ciclo de tratamiento se produjo una toxicidad severa tipo 3 limitante de la dosis, consistente en una mucositis oral aguda. Se observaron otros efectos secundarios, aunque de menor intensidad, entre los que destacaron episodios menores o moderados de mucositis, diarrea, anemia, nausea, fatiga, trombocipenia, neutropenia e hiperlipidemia. Se detectó actividad antitumoral preliminar a todas las dosis ensayadas en el estudio. En estudios en fase II en pacientes con sarcoma, deforolimus mostró una tasa de beneficio clínico del 29%; en otro estudio en cáncer de endometrio la tasa de respuesta fue del 9%, al igual que en neoplasias hematológicas (20). Actualmente se están desarrollando estudios en fase III.
3.3. Inhibidores de EGFR
EGFR o receptor del factor de crecimiento epidérmico es un receptor tirosin-quinasa que se activa por ligandos tales como el factor de crecimiento epidérmico (EGF). Esto desencadena la activación de vías de señalización intracelulares, tales como la proteína-quinasa activada por mitógenos (MAPK) y la fosfatidilinositol 3-quinasa (P13K)-AKT, ambas involucradas en la supervivencia y proliferación celular (22,23). Su sobreexpresión en tumores a menudo se correlaciona con mal pronóstico, disminución de la supervivencia y resistencia a la terapia. La desregulación de EGFR (por su sobreexpresión o por una activación excesiva) contribuye a la proliferación, transformación, angiogénesis, invasión, metástasis y la inhibición de la apoptosis en células tumorales (24). Por lo tanto, moléculas inhibidores de este receptor tienen especial interés como agentes antineoplásicos. Se han identificado dos tipos de inhibidores de la señalización de EGFR:
- Moléculas pequeñas como gefitinib (ZD1839) y erlotinib (OS1774) que inhiben el dominio tirosina quinasa intracelular del receptor EGFR (9).
- Anticuerpos monoclonales, como el cetuximab y matuzumab.
Gefitinib
Gefitinib es un inhibidor tirosin-quinasa selectivo de EGFR de administración oral, ampliamente utilizado en el tratamiento del cáncer de pulmón ya que interrumpe las señales mitogénicas y antiapoptóticos responsables de la proliferación celular, el crecimiento, la metástasis y la angiogénesis. Comparado con agentes antineoplasicos citotóxicos, gefitinib ofrece unas tasas de supervivencia y beneficios equivalentes, pero proporciona una mejor calidad de vida en pacientes con enfermedad avanzada. Sin embargo este beneficio puede variar debido a mutaciones en el receptor EGFR, obteniéndose mejores resultados en mujeres, no fumadores y asiáticos. Gefitinib es bien tolerado y los efectos adversos más comunes, tales como erupciones cutáneas y diarrea, son de gravedad leve, aunque en algunos casos se ha desarrollado enfermedad pulmonar intersticial (ILD). La aprobación de gefitinib como tratamiento de primera elección en Asia desde julio de 2002 para el tratamiento de cáncer de pulmón, se basa en un estudio en fase III que demostró una supervivencia superior, una mayor tasa de respuesta, mejora de la tolerabilidad y significativa mejora de la calidad de vida para los pacientes tratados con gefitinib en comparación con la doble quimioterapia de carboplatino/paclitaxel (25). Sin embargo, desde su aprobación han sido notificados varios casos mortales de ILD. La incidencia de ILD durante el tratamiento con gefitinib varió entre los diferentes grupos étnicos, detectándose la incidencia más alta en la raza japonesa (incidencia acumulada del 4%), mientras que en el resto del mundo fue del 1%. La ILD inducida por gefitinib puede ser mortal en un 30-40% de los casos (26).
Erlotinib
Erlotinib (Tarceva®) es un fármaco de administración oral, inhibidor de la tirosin-quinasa selectivo de EGFR. Estudios in vitro e in vivo, muestran que erlotinib tiene actividad frente a diversos tipos de tumores como colorrectal, pulmón, cabeza, cuello y páncreas. La eficacia antitumoral de erlotinib se ha investigado en clínica como agente único y en combinación con otros agentes. Erlotinib se emplea en cáncer de pulmón tras el fracaso de al menos un régimen de quimioterapia. En un ensayo de fase III, la adición de erlotinib a gemcitabina mejoró la supervivencia en el cáncer de páncreas avanzado. Indicios alentadores de actividad antitumoral también se han observado en varios estudios de fase II en los que se utilizó erlotinib como monoterapia en cáncer de cabeza y cuello, colorrectal, hepatocelular, gastroesofágico biliar y cáncer de ovario. Los efectos adversos detectados en estos estudios fueron hepatitis inducida por medicamentos, enfermedad pulmonar intersticial, síndrome de Stevens-Johnson y necrólisis epidérmica tóxica (27).
Cetuximab
Cetuximab es un anticuerpo monoclonal humanizado que se une con alta afinidad y especificidad al dominio extracelular del receptor del factor de crecimiento epidérmico (EGFR). Esto interfiere con la regulación de la cascada de señalización, lo que conduce a la inhibición de la proliferación celular, angiogénesis y metástasis. Clínicamente, la radioterapia en combinación con cetuximab demuestra una clara ventaja sobre la radioterapia como tratamiento único. Cetuximab potencia la radiosensibilidad de las células tumorales a través de múltiples mecanismos: inhibición de la proliferación de células cancerosas, perturbación del ciclo celular y acumulación de células en fases radiosensibles, e inducción de apoptosis y necrosis. Cetuximab es generalmente bien tolerado, aunque se ha asociado con toxicidad dermatológica incluyendo xerosis, cambios en las uñas, el pelo, telangiectasia o arañas vasculares y erupciones (28). Cetuximab está aprobado para el cáncer de cabeza y cuello, y en 2009 fue aprobado por la FDA para el tratamiento de cáncer colorrectal metastático refractario, ya sea en combinación con irinotecán o como monoterapia (29), dicha aprobación recoge que este fármaco sólo puede usarse en aquellos casos en los que no exista una mutación en el gen KRAS. Este gen codifica una pequeña proteína G de la ruta de EGFR, de tal forma que en 2012, la FDA, aprobó un test genético para detectar de forma rutinaria siete tipos de mutaciones en el gen KRAS, permitiendo así dirigir de forma adecuada el tratamiento antineoplásico. Este efecto se detecta exclusivamente para el tratamiento del cáncer colorrectal y también se produce con otros inhibidores de EGFR.
Matuzumab
Matuzumab es un anticuerpo monoclonal humanizado IgG1 que se une a EGFR con alta afinidad. Matuzumab ha sido evaluado en cáncer de cabeza y cuello, gástrico, colorrectal, esofágico, cervical y de pulmón. Un estudio preclínico en líneas celulares muestra que aunque matuzumab se une de manera eficiente a EGFR y bloquea su fosforilación, no es tan eficaz como cetuximab a la hora de inhibir la proliferación de células tumorales. Estudios en fase II de matuzumab en mujeres con cáncer de ovario resistente a platinos muestra que el fármaco tiene un perfil de toxicidad favorable, observándose erupción acneiforme, erupción cutánea, náuseas, vómitos y diarrea aunque con una incidencia más baja que la obtenida con cetuximab (30).
3.4. Inhibidores de la tubulina
Existe una gran cantidad de moléculas capaces de unirse a la tubulina e interferir en el mecanismo de formación de los microtúbulos, de manera que las células detienen su ciclo celular y desencadenándose la apoptosis. Los compuestos que modulan la actividad de tubulina pueden dividirse de forma general en dos grandes grupos: los inhibidores de su polimerización, como la colchicina y la vincristina, que se unen a ésta impidiendo que forme microtúbulos; y los agentes estabilizantes de microtúbulos, como el paclitaxel y el docetaxel, que se unen preferentemente a la tubulina ensamblada, minimizando la disociación de la tubulina-GDP de los extremos de los microtúbulos e induciendo el ensamblaje de la tubulina-GDP normalmente inactiva. El éxito clínico del paclitaxel y el docetaxel ha conducido a la búsqueda de nuevos compuestos con el mismo mecanismo de acción y al descubrimiento en los últimos años de una gran cantidad de agentes estabilizantes de microtubulos con al menos dos sitios de unión distintos.
DJ-927
DJ-927 es un nuevo taxano que tiene como ventajas su alta hidrosolubilidad (elimina la necesidad de emplear vehículos tóxicos, minimizando el riesgo de reacciones de hipersensibilidad), su elevada biodisponibilidad oral y una potente actividad antitumoral. Su mecanismo de acción implica la inhibición de la tubulina lo que causa detención de la división de ADN en la célula y apoptosis. Este compuesto exhibió una citotoxicidad mayor que los taxanos existentes tales como paclitaxel y docetaxel contra diversos tipos de tumores, especialmente en modelos de cáncer colorrectal. En particular, DJ-927 muestra una marcada eficacia en ensayos, in vitro e in vivo, con células tumorales que presentan una la resistencia intrínseca o adquirida a taxanos desarrollada por expresión de bombas de eflujo como la glicoproteína P (P-gp). En estudios en fase II como tratamiento de segunda línea para pacientes con cáncer colorrectal tras fallo de irinotecán u oxaliplatino, y en pacientes con cáncer gástrico avanzado que no han respondido a 5-fluoruracilo, el tratamiento fue bien tolerado, alcanzándose una respuesta positiva en el 40% de los casos (31).
ABT-751
ABT-751 es un agente citotóxico novedoso, de administración oral, que se une a la tubulina e inhibe la polimerización de los microtúbulos, lo que conduce a un bloqueo en la fase G2/M en el ciclo celular y a la apoptosis celular. ABT-751 inhibe la proliferación de diversas líneas celulares humanas derivadas de tumores, incluyendo los fenotipos resistentes a paclitaxel y doxorubicina (32). Una vez administrado, ABT-751 se absorbe rápidamente alcanzándose la concentración máxima (Cmax) a las 3 h y con una semivida de 4,4 a 16,6 h. La toxicidad producida por ABT-751 fue determinada en un estudio en fase I, detectándose neuropatía periférica, estreñimiento, fatiga y mialgia. ABT-751 está actualmente en ensayos fase II para el cáncer de mama recurrente, cáncer renal, cáncer colorrectal y de pulmón.
EPO-906
EPO-906 (epotilona B) es un miembro de una nueva clase de agentes estabilizantes de microtúbulos conocidos como las epotilonas. EPO906 promueve la polimerización de los heterodímeros de tubulina y estabiliza los microtúbulos contra la despolimerización, provocando la detención del ciclo celular mitótico y la muerte celular por apoptosis (32). En estudios preclínicos, EPO906 muestra actividad anticancerígena tanto in vitro como in vivo contra varios tipos de cáncer, incluyendo modelos resistentes a paclitaxel. Es importante destacar que, EPO906 actúa sobre células que han desarrollado resistencia por sobre-expresión de glicoproteína-P o bomba de eflujo. En estudios clínicos en fase I se observó que el efecto tóxico limitante de dosis fue la diarrea. Los ensayos se realizaron en pacientes con cáncer de colon, mama, pulmón y ovario, incluyendo pacientes que habían recibido taxanos previamente. Posteriormente, se realizaron dos estudios en fase II en monoterapia para cáncer de ovario, próstata, mama y cáncer renal, observándose respuesta antitumoral en todos los casos. En consecuencia, los estudios preclínicos y clínicos indican que EPO906 tiene un espectro relativamente amplio de actividad, incluyendo los tumores resistentes a paclitaxel.
TZT-1027
TZT-1027 es un derivado de dolastatina-10, que fue seleccionado por su alta actividad antitumoral in vitro. TZT-1027 se desarrolló como un nuevo agente inhibidor de la polimerización de la tubulina mediante su unión a los microtúbulos, impidiendo la división celular y activando la apoptosis de las células tumorales, como los alcaloides de la vinca y los taxanos, pero los experimentos in vitro mostraron que además de este efecto de citotoxicidad directa, era capaz de inhibir la angiogénesis del tumor, una actividad que no se ve con los alcaloides vinca o taxanos (33,34). En un estudio en fase I se pudo determinar que la toxicidad limitante de dosis fue debida a neutropenia, neuropatía periférica, leucopenia y fatiga. Los resultados de un estudio en fase II desarrollado en 32 pacientes con cáncer de pulmón mostraron un aumento en la supervivencia global de las pacientes (35).
3.5. Otros agentes inductores de apoptosis
Fenoxodiol
Fenoxodiol es una isoflavona sintética que ha demostrado actividad antitumoral frente a cáncer de ovario, y que se encuentra en ensayos en fase I para cáncer de próstata y de cérvix. A pesar de la progresión en los ensayos clínicos, la diana sobre la que actúa fenoxodiol no está muy clara, más bien, se cree que regula múltiples señales de transducción. Estudios mecanísticos han demostrado que fenoxodiol inhibe la ADN topoisomerasa II e induce la apoptosis. Esta apoptosis puede desencadenarse de forma dependiente o independiente de caspasas en función del tipo celular. Así se ha descrito un mecanismo dependiente de caspasas en las células de cáncer de ovario, a través de la vía de transducción de señal de Akt (36), mientras que en otros tipos de cáncer (cabeza, cuello y colon) fenoxodiol induce apoptosis independiente de caspasas por expresión de p21WAF1 (37). Otro mecanismo por el que se cree que actúa fenoxodiol es por alteración del ciclo redox esencial de las células. El control del equilibrio redox intracelular es crítico para la viabilidad celular, de manera que la perturbación de los principales pares redox tales como la relación NADH/NAD+ o el glutatión puede tener profundos efectos sobre las células desencadenándose la apoptosis.
La mayoría de los casos de cáncer de ovario con quimioresistencia a platino y taxanos se asocian a la sobreexpresión de factores antiapoptóticos. Fenoxodiol, in vitro, induce apoptosis en células de cáncer de ovario quimioresistentes y restaura la sensibilidad a la quimioterapia de platino, taxanos y topotecan en células quimioresistentes. Estos datos apoyan el estudio en fase II de fenoxodiol llevado a cabo en 40 mujeres con cáncer de ovario quimiorresistente. El fenoxodiol se administró en terapia combinada con taxanos o paclitaxel, y los resultados indicaron que la administración de fenoxodiol da lugar a mayores tasas de respuesta entre las mujeres con cáncer de ovario quimioresistente.
Inhibidores de c-Kit y PDGFR
c-kit (receptor del factor de células madre) y PDGFR (receptor del factor de crecimiento derivado de plaquetas) son receptores tirosin-quinasa que estimulan el crecimiento de la célula tumoral, la angiogénesis y la vasculogénesis. c-Kit y PDGFR, así como sus respectivos ligandos están sobre-expresados en un 70% de los cánceres de ovario (38). Las mutaciones de c-kit en las células intersticiales de Cajal en el tracto digestivo son probablemente la clave del 85% de los tumores del estroma gastrointestinal, ya que estas mutaciones desencadenan cascadas de señalización que promueven la proliferación celular y supervivencia. Las mutaciones de PDGFR están implicadas en un 5% de estos tumores.
Imatinib (STI-571, Gleevec®) es una molécula pequeña inhibidora de c-kit, PDGFRA, PDGFRB y de la proteína de fusión BCR-ABL, que ha revolucionado el pronóstico de los pacientes con tumor del estroma gastrointestinal, con un beneficio clínico en el 85% de los casos. Sin embargo un alto porcentaje de los pacientes tratados con imatinib acaban desarrollando resistencia (10-20% muestran resistencia natural y un 50% resistencia adquirida), lo que ha llevado a la necesidad de emplear imatinib en combinación con otro agente antineoplásico con el fin de superar las resistencias a este fármaco. En este sentido, se ha observado que su combinación con un agente proapoptótico potencia la muerte celular y previene de la resistencia a imatinib (39). Esto ha llevado a nuevos estudios sobre el mecanismo de acción de imatinib, determinándose que induce apoptosis en una serie de tumores como leucemia, leucemia mielógena, estroma gastrointestinal, cáncer ganglionar de la retina y glioblastoma. La apoptosis se manifiesta a través de la fragmentación nuclear, la degeneración de la membrana mitocondrial y la activación de las caspasas.
4. CONCLUSIÓN
La búsqueda de nuevas dianas para el tratamiento de tumores pretende, por un lado una mayor eficacia en la eliminación o detención del crecimiento tumoral y por otro evitar o dificultar la aparición de resistencias. Los nuevos agentes antineoplásicos con actividad proapoptótica, han demostrado en diversos ensayos clínicos en los que se ha evaluado como monoterapia que son capaces de disminuir el tamaño de la masa tumoral en diferentes tipos de cáncer, incluso en tumores resistentes a otros tratamientos; pero también se ha demostrado que muchos de ellos ofrecen un efecto sinérgico al utilizarse en combinación con los agentes antineoplásicos actuales evitando incluso la resistencia de las células tumorales hacia estos últimos.
5. AGRADECIMIENTOS
Trabajo financiado por la Consejería de Educación de la Comunidad de Madrid y el Fondo Social Europeo mediante el Plan Regional de Investigación Científica e Innovación Tecnológica; y por el Programa de Grupos de Investigación Santander-UCM GR35/10, Grupo: Administración parenteral de medicamentos.
6. REFERENCIAS
1. Chu, E.; DeVita VT. (2001) Principles of Cancer Management: Chemotherapy. Cancer, Principles and Practice of Oncology, 6th Edition. Philadelphia; p 289-386.
2. Goldie, JH.; Coldman, AJ. (1979) A mathematic model for relating the drug sensitivity of tumors to their spontaneous mutation rate. Cancer treat Rep 63, 1727-1731.
3. Skipper, HE.; Simpson-Herren, L. (1985) Relationship Between Tumor Stem Cell Heterogeneity and Responsiveness to Chemotherapy. Important Advances in Oncology. Philadelphia; p 63-77.
4. Arends, MJ.; Wyllie, AH. (1991) Apoptosis: Mechanisms and roles in pathology. Int Rev exp Pathol 32, 223-254.
5. Wyllie, AH. (1992) Apoptosis and the regulation of cell numbers in normal and neoplasic tissues: an overview. Cancer Metast Rev 11, 95-103.
6. Gershenson, LE.; Rotello, RJ. (1992) Apoptosis: a different type of cell death. FASEB J 6, 2450-2455.
7. Parchment, RE. (1993) The implications of a unified theory of programmed cell death, polyamines, oxyradicals, and histogenesis in the embryo. Int J Dev Biol 37, 75-83.
8. Sachs, L.; Lotem, J. (1993) Control of programmed cell death in normal and leukemic cells: new implications for therapy. Blood 82, 15-21.
9. Agarwal, R.; Linch, M.; Kaye, SB. (2006) Novel therapeutic agents in ovarian cáncer. Eur J Surg Oncol 32, 875-886.
10. Satyam, A.; Hocker. MD.; Kane-Maguire, KA.; Morgan, AS.; Villar, HO.; Lyttle, MH. (1996) Design, synthesis, and evaluation of latent alkylating agents activated by glutathione S-transferase. J Med Chem 39(8), 1736-1747.
11. Lyttle, MH.; Satyam, A.; Hocker, MD.; et al. (1994) Glutathione-Stransferase activates novel alkylating agents. J Med Chem 37(10), 1501-1507.
12. Thévenin, AF.; Zony, CL.; Bahnson, BJ.; Colman, RF. (2011) GSTpi modulates JNK activity through a direct interaction with JNK substrate, ATF2. Protein Sci 20(5), 834-848.
13. Vergotea, I.; Finklerb, N.; Del Campoc. J. et al. (2009) Phase 3 randomised study of canfosfamide (Telcyta®, TLK286) versus pegylated liposomal doxorubicin or topotecan as third-line therapy in patients with platinum-refractory or –resistant ovarian cancer. Eur J Cancer 45, 2324-2332.
14. Turella, P.; Filomeni, G.; Dupuis, ML. et al. (2006) A strong glutathione S-transferase inhibitor overcomes the p-glycoproteinmediated resistance in tumour cells. 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) triggers a caspasedependent apoptosis in MDR1-expressing leukemia cells. J Biol Chem 281, 23725-23732.
15. Filomeni, G.; Turella, P.; Dupuis, ML. et al. (2008) 6-(7-nitro-2, 1, 3-benzoxadiazol-4-ylthio)hexanol, a specific glutathione Stransferase inhibitor, overcomes the multidrug resistance (MDR)-associated protein 1-mediated MDR in small cell lung cancer. Mol Cancer Ther 7, 371-379.
16. Pellizzari Tregno, F.; Sau, A.; Pezzola, S. et al. (2009) In vitro and in vivo efficacy of 6-(7-nitro-2, 1, 3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) on human melanoma. Eur J Cancer 45, 2606-2617.
17. Tentori, L.; Dorio, AS.; Mazzon, E.; Muzi, A.; Sau, A.; Cuzzocrea, S. et al. (2011) The glutathione transferase inhibitor 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) increases temozolomide efficacy against malignant melanoma. Eur J Cancer 45, 1219-1230.
18. Huang, S.; Shu, L.; Easton, J. et al. (2004) Inhibition of Mammalian Target of Rapamycin Activates Apoptosis Signal-regulating Kinase 1 Signaling by Suppressing Protein Phosphatase 5 Activity. J Biol Chem 279(35), 36490-36496.
19. Temkin, SM.; Yamada, SD.; Fleming, GF. (2010) A phase I study of weekly temsirolimus and topotecan in the treatment of advanced and/or recurrent gynecologic malignancies. Gynecol Oncol 117, 473-476.
20. Yap, TA.; Garrett1, MD.; Walton, MI.; Raynaud, F.; De Bono, JS.; Workman, P. (2008) Targeting the PI3K–AKT–mTOR pathway: progress, pitfalls, and promises. Curr Opin in Pharmacol 8, 393-412.
21. Strimpakos, AS.; Karapanagiotou, EM.; Saif, MW.; Syrigos, KN. (2009) The role of mTOR in the management of solid tumors: An overview. Cancer Treat Rev; 35:148-159.
22. Ciardiello F. (2005) Epidermal growth factor receptor inhibitors in cancer treatment. Future Oncol 1(2), 221-234.
23. Salomon, DS.; Brandt, R.; Ciardiello, F. et al. (1995) Epidermal growth factorrelated peptides and their receptors in human malignancies. Crit Rev Oncol/Hematol 19, 183-232.
24. Dummer Meira, D.; Nóbrega, I.; De Almeida, VH.; Mororo, JS.; Cardoso, AM.; Silva, R. (2009) Different antiproliferative effects of matuzumab and cetuximab in A431 cells are associated with persistent activity of the MAPK pathway. Eur J Cancer 45, 1265-1273.
25. Oh, IJ.; Jung-Ban, H.; Kim, KS.; Kim, YC. (2012) Retreatment of gefitinib in patients with non-small-cell lung cancer who previously controlled to gefitinib: A single-arm, open-label, phase II study. Lung Cancer 77, 121-127.
26. Chang, SC.; Chang, CY.; Chang, SJ.; Yuan, MK.; Lai, YC.; Liu, YC. (2013) Gefitinib-Related Interstitial Lung Disease in Taiwanese Patients With Non–Small-Cell Lung Cancer. Clin Lung Cancer 14(1), 55-61.
27. Luo, Q.;, Gu, Y.; Zheng, W. et al. (2011) Erlotinib inhibits T-cell-mediated immune response via down-regulation of the c-Raf/ERK cascade and Akt signaling pathway. Toxicol Appl Pharmacol 251, 130-136.
28. Duffour, J.; The´zenas, S.; Dereure, O. et al. (2010) Inter-observer agreement between dermatologists and oncologists in assessing dermatological toxicities in patients with metastatic colorectal cancer treated by cetuximab-based chemotherapies: A pilot comparative study. Eur J Cancer 46, 3169-3174.
29. Dinh, P.; Harnett, P.; Piccart-Gebhart, MJ.; Awada, A. (2008) New therapies for ovarian cancer: Cytotoxics and molecularly targeted agents. Crit Rev Oncol/hematol; 67:103-112.
30. Seiden MV, Burris HA, Matulonis U, et al. A phase II trial of EMD72000 (matuzumab), a humanized anti-EGFR monoclonal antibody, in patients with platinum-resistant ovarian and primary peritoneal malignancies. Gynecol Oncol 104, 727-731 (2007).
31. Ismael. GF.; Dornelles. D.; Mano, MS.; Awada. A. (2008)Novel cytotoxic drugs: Old challenges, new solutions. Cancer Treat Rev 34, 81-91.
32. Hotta, K.; Ueok, H. (2005) New cytotoxic agents: a review of the literature. Crit Rev Oncol/hematol 55, 45-65.
33. Natsume, T.; Watanabe, J.; Koh, Y. et al. (2003) Antitumor activity of TZT-1027 (soblidotin) against vascular endothelial growth factor secreting human lung cancer in vivo. Cancer Sci 94, 826-833.
34. Natsume T, Watanabe J, Koh Y, et al. (2003) Antitumor activity of TZT-1027 (Soblidotin) against vascular endothelial growth factor-secreting human lung cancer in vivo. Cancer Sci 94, 826- 833.
35. Riely, GJ.; Gadgeel, S.; Rothman, I.; Saidman, B.; Sabbath, K.; Feit, K. (2007) A phase 2 study of TZT-1027, administered weekly to patients with advanced non-small cell lung cancer following treatment with platinum-based chemotherapy. Lung Cancer 55, 181-185.
36. Kamsteeg, M.; Rutherford, T.; Sapi, E.; et al. (2003) Phenoxodiol –an isoflavone analog– induces apoptosis in chemoresistant ovarian cancer cells. Oncogene 22(17), 2611-2620.
37. Aguero, MF.; Facchinetti, MM.; Sheleg, Z.; Senderowicz, AM. (2005) Phenoxodiol, a novel isoflavone, induces G1 arrest by specific loss in cyclin-dependent kinase 2 activity by p53- independent induction of p21WAF1/CIP1. Cancer Res 65(8), 3364-3373.
38. Tonary, AM.; Macdonald, EA.; Faught, W. et al. (2000) Lack of expression of c-Kit in ovarian cancers is associated with poor prognosis. Int J Cancer 89(3), 242-250.
39. Reynoso, D.; Nolden, LK.; Yang, D.; et al. (2011) Synergistic induction of apoptosis by the Bcl-2 inhibitor ABT-737 and imatinib mesylate in gastrointestinal stromal tumor cells. Mol Oncol 5, 93-104.